# ExploreInformatics: Part 2
Welcome to Part 2 of ExploreInformatics! In this project, you will train and test a hidden markov model to detect motif occurrences in a sequence.
## Setup
Before starting this portion of the assignment, enter your `cs181-projects24` directory and run `git pull`. This will pull the necessary scripts and support files for this assignment into your directory. Then enter the cs1810 docker container by running `./run-docker`. All of the packages needed for this application should already be embedded into the Docker image.
All of the code for this project can be found in the `motif_hmm` directory in the `cs181-user` directory. Everything you will need to complete this assignment can be found within that folder.
In particular, you will implement some minor preprocessing of the data we have given you in `preprocessing.py`. From here, you will implement the training and testing of your HMMs in `hmm_traintest.py`. Finally, you will make minor edits to `explore_hmm.py` and run the script to see the results of your implementations (i.e., `explore_hmm.py` is where you will actually call the functions in the other two files).
:::danger
You need to run `pip install numpy==1.26 --break-system-packages` to be able to use the docker container.
:::
:::spoiler Docker Issues? Want to use conda/pip instead?
The only required packages (that would need to be installed) are `hmmlearn` and `numpy==1.26`. Instead of using the Docker container, you can manually install these packages into a conda environment or through `pip3`. Let us know if you would like to do this and do not know how to!
:::
\
Here is a quick guide through the files in this project. There are labeled TODOs in all three files.
| **File** | **Description** |
|--------------------|-----------------------------------------------------------------------------------------------------------------------|
| `explore_hmm.py` | Overarching file to test your code. Run `python3 explore_hmm.py` to run the HMMs as you progress through the project. |
| `hmm_traintest.py` | Contains the train and test functions for the HMM that you will implement. |
| `preprocess.py` | Contains the preprocessing steps for the FASTA files. Will also prepare and package data for training/testing. |
| `data/` | Contains the FASTA files for four TFs and the mystery dataset. |
## Biological Background
Motifs, in the realm of transcription factors, are short, specific sequences within DNA that serve as binding sites for these regulatory proteins. They act as key points where transcription factors attach, influencing gene expression and the subsequent production of RNA from DNA.
Creating models capable of detecting motif occurrences is crucial in deciphering gene regulation. By identifying these motifs, scientists gain insights into the complex mechanisms governing gene expression. Understanding where and how transcription factors bind helps unravel the intricacies of cellular processes, disease development, and evolutionary patterns.
These motifs, unique to specific factors, can be anywhere from 6-20 nucleotides in length. At each position of a motif, there is a different likelihood of observing a specific nucleotide. As a result, there is no one exact way that a motif might be found in the genome. For this reason, probabilistic models like HMMs are simple but effective methods for motif recognition.
If you are interested in reading a more thorough introduction to motifs, here is an [introductory resource from UCSC](https://bio.libretexts.org/Bookshelves/Computational_Biology/Book%3A_Computational_Bio[…]02%3A_Introduction_to_regulatory_motifs_and_gene_regulation).
:::spoiler What does a motif look like?

Notice how at each position certain nucleotides are more likely to be observed. In the corresponding visualization, that nucleotide is then larger at that position.
:::
### Why HMMs?
Hidden Markov Models are excellent candidates for recognizing patterns in sequential data. In this case, we can hypothesize that there are underlying states in the DNA: motif or background. We can think of the background state producing emissions related to other genomic features that are **not** our motif. Then, at each position, we want to predict if the observed emission (nucleotide) came from the motif or background state!
It is important to note that training an HMM is an unsupervised process. This means that the training process utilizes unlabeled data and the [Expectation-Maximization algorithm](https://en.wikipedia.org/wiki/Expectation–maximization_algorithm). This process makes the assumption that there is some underlying state that the model should detect within our observations.
### What's Our Training Data?
All of the data for this project comes from the [Fly Factor Survey](https://pgfe.umassmed.edu/ffs/) which is a database of *Drosophila* transcription factor DNA-binding specifities. This database includes the motif information for hundreds of *Drosophila* transcription factors and the raw DNA sequences that were used to derive them.
In the `data` directory within the `motif_hmm` directory, you will find four motif files for *Drosophila* transcription factors Chinmo, Ken Barbie, Lame Duck, and Sugarbabe. Yes, these are real names of *Drosophila* transcription factors. In this project, you will write code that is able to detect the motifs of each of these transcription factors based on the provided data!
:::spoiler **Why do we only train on positive sequences?**
You might notice that we only train our model on positive samples. EM is an unsupervised process - there are no labels provided to `hmmlearn`! A positive sequence, say of length 30, will contain a motif of around length 10. That means that 1/3 of the underlying states for this sequence are 1 (motif) and the remaining 2/3 are 0 (background).
So, within our positive data samples themselves, we are providing the model with ample background sequences. The magic of E-M is that we tell it that there are two hidden states, and a probabilistic model that takes advantage of this will perform the best!
:::
:::spoiler **[Important] Sequence Logos**
These images will be helpful towards the end of the assignment! Here are the motif logos or visualized PWMs for each of the four transcription factors. Note that the height and size of a given letter at a given position reflects a) the frequency of that letter at that position, among all sequences containing the motif and b) the information content of the position itself, also among all sequences with the motif.
*Chinmo*
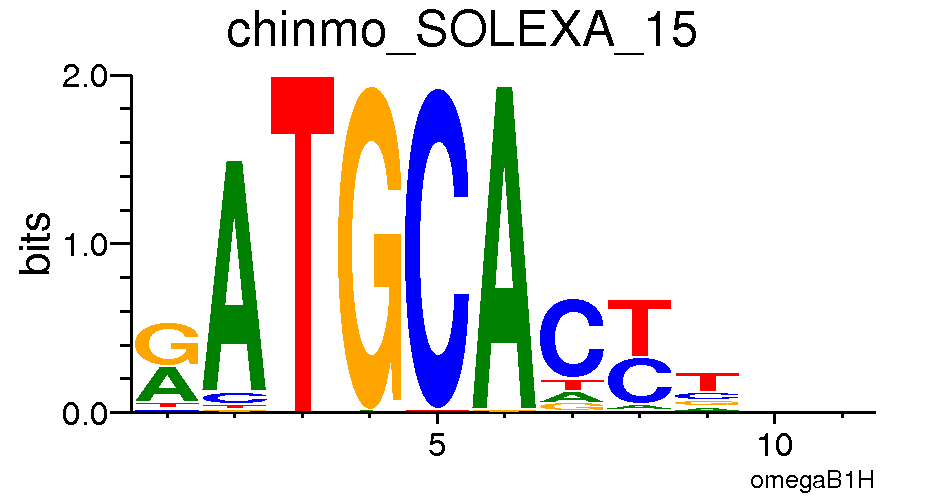
*Ken Barbie*
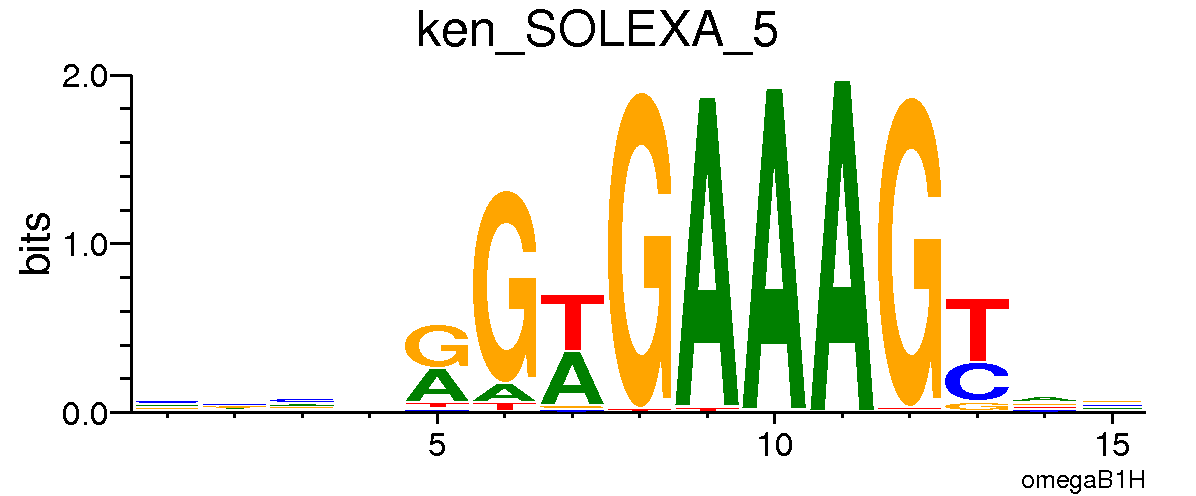
*Lame Duck*
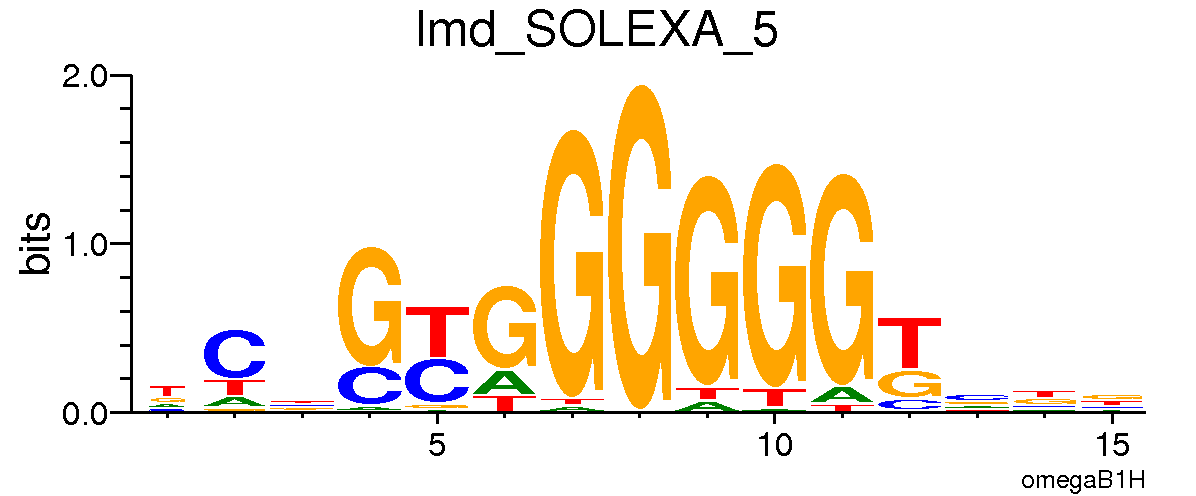
*Sugarbabe*
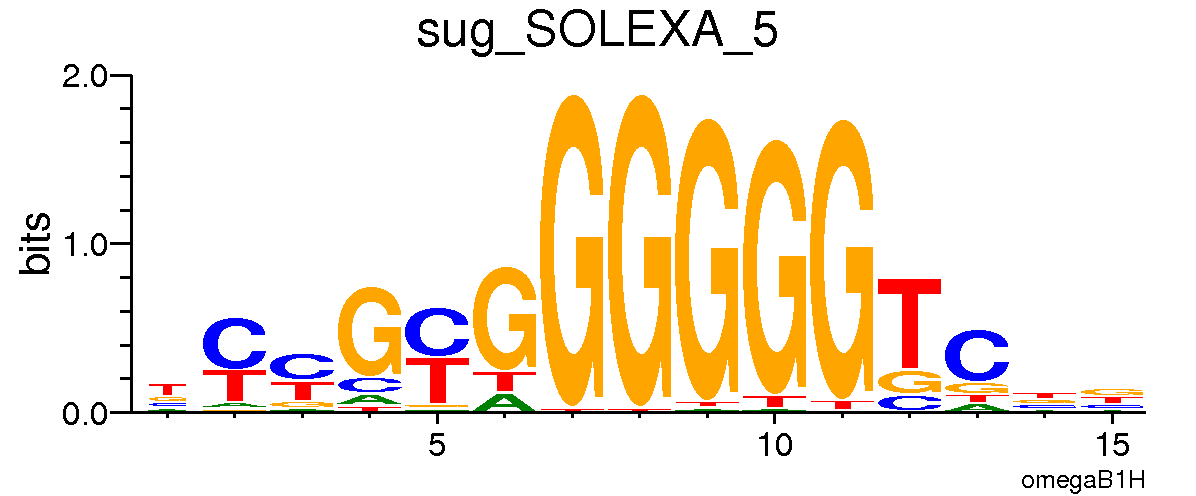
:::
## Preprocessing
Before we can actually train the model, we need to preprocess the data so that it is in the correct format for training. This, more often that not, is the most important step in utilizing models like HMMs or deep learning frameworks.
Open `preprocess.py` to complete the following tasks. While a majority of the code is already provided in this section, we encourage you to think about what each of the functions are doing!
All of the data that we have provided you is *in vivo* samples of motif occurrences, meaning this data was all generated via experimentation on living organisms. However, we need negative/noise samples. Sometimes, this type of data is created *in silico*, meaning the data is generated according to some simulation, rather than a real-life experiment.
:::success
**Task 1a**: Fill out `gen_negatives` to generate a list of negative sequences.
:::
Now, you need to be able to read in the positive data in the `*.fa` (FASTA) or sequence files. Make sure to observe the format in which the data is provided. When you are collecting your data, you only want **the DNA sequences**.
:::success
**Task 1b**: Fill out `read_motif_file` to gather the positive samples from the FASTA files.
The input `fname` is the filepath to some arbitrary FASTA file. The output should be a list of strings where each string is a sequence from the FASTA file.
:::spoiler Hint!
Using a while loop to iteratively process the file will be very helpful! Consider, also, how to skip rows with unnecessary information.
:::
## `hmmlearn`: Training and Testing
Now, we can begin to actually train our HMM! In order to do so, start by opening up `hmm_traintest.py`. Within `hmm_traintest.py`, you will complete the main set of tasks for this project, which begins with training.
### Training
In order to get started on training your HMM, you will need to start by specifying the *initial* parameters in your model in the `train_hmm` function. From here, you will need to pass these parameters into the instantiation of a CategoricalHMM, which we have given you in the stencil code. The CategoricalHMM will start from these parameters when training, and learn to vary them slightly to maximize the probability of observing the sequences we train it on. *This means that, if you select illogical initial parameters, the model performance will likely reflect them.* Within a reasonable range for the different parameters, play around and see how model performance varies. In order to see what you might need to execute this step, we recommend looking at the following [documentation](https://hmmlearn.readthedocs.io/en/latest/api.html#hmmlearn.hmm.CategoricalHMM).
We recommend setting the number of iterations for your HMM to be around 100, to avoid overfitting. Feel free to play around with the value, but be wary that overfitting or underfitting the HMM may lead to subpar results.
:::success
**Task 2a**: Determine and instantiate your parameters for the CategoricalHMM.
**Edit:** When you are setting the parameters, **do not** do it within the the model initialization. Instead, set the parameters separately after model initialization. For example ` model.startprob_ = ...`
:::spoiler Hint!
We've given you the two states we expect for this model – motif and background. Which of these do you think should be more common if motifs tend to be a fair amount shorter than the sequences that contain them? With this in mind, what might the initial probability distribution look like?
Additionally, note that we test our motif HMMs by seeing if a given sequence has *any* motif states in its most likely sequence - as predicted by the model. What might happen to our predictions if we gave the motif state a high start probability?
In general, what will happen if we initialize our distributions so that it's fairly easy to reach a motif state?
:::
Once you have decided on your initial parameters for the model, it's time to start fitting! The first step to fitting will be finding the data you will use to fit the model. Remember that `train_hmm` takes in the name of a motif and a file containing training data for each motif that we originally preprocessed. In particular, the training data is shaped as a dictionary that keys on motif names and returns a tuple of sequences and labels.
:::success
**Task 2b**: In `train_hmm`, read in the relevant training data from the function inputs.
:::
Once you have figured out how to access the set of training sequences for the given motif, look through the [documentation](https://hmmlearn.readthedocs.io/en/latest/api.html#hmmlearn.hmm.CategoricalHMM) for CategoricalHMMs once more. In particular, look at the fit function. How can you convert a list of sequences into data format required to fit a CategoricalHMM?
:::success
**Task 2c**: Within `train_hmm`, convert the motif data (determined by the motif and training file passed in) into a format that our CategoricalHMM can handle. You might want to make this conversion step a helper function, so that you can use it later during testing. Then, fit your model.
:::spoiler Hint!
At some point in the process of converting your motif sequences into the format required by CategoricalHMM, you will have to convert your DNA strings into numbers. What data structure could help you to do this efficiently?
:::
Now that you have sucessfully devised a way to train models by motif, it's time to actually train!
:::success
**Task 2d**: Fill out the training TODO in `explore_hmm.py` in order to populate the trained_models dictionary for each motif.
:::
In order to test that your training is working, try commenting out the testing steps (everything below the training step in `explore_hmm.py`) and ensure that the script runs.
### Testing
In order to make sure your models in `explore_hmm.py` are working seamlessly, you will need to devise a way to test them. In particular, you will need to fill in `test_hmm` in `hmm_traintest.py`.
`test_hmm` takes in a singular model, a list of DNA sequences to test, and a potential list of ground truth labels for each sequence. In order to evaluate the model taken in, you will need to decode the list of sequences passed in for testing.
:::success
**Task 3a**: Use the `decode` function in order to get back the most likely sequence of states for each observed sequence.
:::spoiler Hint!
This function might require you to put your data into a similar format as `fit`.
:::
Once you have decoded your observations, you will need to manipulate the sequence of states returned. In particular, `decode` returns a 1D array (of size equal to the total number of nucleotides across all sequences) of most likely states. However, we want to evaluate each sequence individually.
:::success
**Task 3b**: From the output of `decode`, extract the most likely sequence of states for each observation (separately).
:::
Once you have this list of states for each observed sequence, you will need to determine whether the model predicted that the sequence contained the motif or not. In order to do so, you will need to iterate over each list of states from `decode` and determine whether the model predicted that the associated sequence reached at least one motif state or not.
:::success
**Task 3c**: In the `test_results` variable, mark all sequences that reach a motif state with a 1, and all other sequences with a 0.
:::
Once you have this list of test results, you will need to evaluate how many positives there were, or whether they match some set of ground truths.
:::success
**Task 3d**: If ground truth labels are available, calculate the percent of observations in `test_results` that match with their ground truth labels. Otherwise, compute the percent of observations that are positive for the motif on which we trained the inputted model.
:::
To test that `test_hmm` is working, make sure to run `explore_hmm.py` up to the point where we introduce mystery data. Don't worry too much if the test accuracy isn't close to 100%; since the algorithm is unsupervised, we don't expect anyone to achieve such accuracy.
Note that you have programmed `test_hmm` such that when you run `explore_hmm` up to the mystery bag section, the test output will be *with* ground truths from the test set. As a result, the scores you see will reflect the *accuracy* of your model.
Later, when you run `explore_hmm` on the mystery bag of sequences, there will be no ground truth. In this case, the output of `test_hmm` will be *the percentage of sequences that are positive for the motif associated with the model.* This distinction is critical for evaluating your models, as well as making inferences based on the mystery bag results.
:::success
**Task 3e**: Run `explore_hmm.py` to verify that your test methods are working.
:::
With the `train_hmm` and `test_hmm` functions filled out, you can begin applying your trained models!
## Applying Your Models
Now that you have trained your four models, we want to put them to good use! In the `data` directory, we have provided a mystery pickle file. (We are now entering a realm of complete biological wildness).
For the sake of sticking with the theme, let us imagine that *Drosophila* actually relates to a dinosaur and not a fruit fly. Pranav was recently taking a walk in India Point Park when he stumbled across the skeletal remains of what looked like a dinosaur *Drosophila* skeleton. After conducting some tests, he found some DNA sequences that were mysteriously preserved. He wanted to determine what motifs were present in order to better understand what transcription factors might have played a role.
Using your code from the last two components, you will apply these trained HMMs on the mystery bag to see which of the TFs' motifs are enriched. In the previous section, we assessed accuracy of our models using ground truth labels. In this case, we have no labels, so, we need to run each of our HMMs on all of the sequences and see what proportion of sequences yielded a positive prediction of a motif.
With this information for each HMM, we should be able to predict which TFs might be involved in this strange phenomena - even if the accuracy of our models was not perfect!
:::success
**Task 4**: Uncomment the mystery bag code and run the code on the mystery dataset. In **application.pdf**, present which transcription factors you think were involved. Make sure to discuss why **each factor** could have been or could not have been involved.
Once you have made your predictions, discuss *how confident you are about the makeup of your bag* (what portion of the samples are from each TF involved in the mystery bag). Consulting the **sequence logos** will likely be helpful here.
:::spoiler Hint!
There are **two** transcription factors in the mystery bag. If you are seeing strange results, tweaking your initializations for the transition probabilities and start states will help.
:::
:::success
**Extra Credit:** Make a plot or two that summarize your results from testing/training and the mystery bag!
:::
## Wrapping Up
Now, you have successfully trained your HMMs and applied them to a very hypothetical scenario. If you want to learn more about the E-M algorithm/want to implement the code for `hmmlearn` yourself, consider taking CS182! Before you are done, please answer the following question in **application.pdf**.
:::success
**Task 5**: Was there a transcription factor whose corresponding HMM was not as effective or accurate? Regardless of whether you made this observation, provide **3 reasons** you think that this could occur.
:::spoiler **Hints**
* We are not asking for anything super detailed! Think about what assumptions our training setup and the HMMs make.
* It might help to consult the sequence logos again!
:::
:tada: :tada: :tada: Congratulations, you have finished your last project for CS1810! :tada: :tada: :tada:
:::warning
**Task 6**: In your **application.pdf**, please leave any feedback you might have so we can make future iterations of the course better. We really appreciate it! :smile:
:::
Developed by pmahable and cbaker20, 2023. Revised by pmahable in 2024.







