[TOC]
# Annotate the eukaryotic and bacterial taxonomy of a metagenome using sourmash
C. Titus Brown
Aug 5, 2025
DRAFT tutorial; updates &c will happen here: [sourmash#3765](https://github.com/sourmash-bio/sourmash/issues/3765).
## Description
This tutorial demonstrates how to use sourmash to search a metagenome signature against a combined database of GTDB+NCBI eukaryote genomes. This addresses a challenge in metagenomics where people may want to use the GTDB database of genomes and/or the GTDB taxonomy, but also want to discover eukaryotic organisms that are not in GTDB.
At the end, this tutorial provides several visualizations of taxonomic composition as well.
## Compute requirements
You'll need about 70 GB of disk space to run this tutorial, including the software install. The memory and CPU requirements are minimal (1 GB RAM, ~2 minutes on one CPU).
## Install sourmash, taxburst, and the betterplot plugin for sourmash
Install [sourmash](https://sourmash.readthedocs.io/), [taxburst](https://taxburst.github.io/taxburst/), and [sourmash betterplot](https://github.com/sourmash-bio/sourmash_plugin_betterplot/).
Create a new conda environment with sourmash installed:
```
conda create -n smash-euk-tutorial -y 'sourmash >= 4.9.3' python=3.12
```
Activate, and then install some pure Python packages:
```
conda activate smash-euk-tutorial
pip install sourmash_plugin_betterplot taxburst
```
## Download databases
We need two of the [sourmash indexed RocksDB databases](https://sourmash.readthedocs.io/en/latest/databases.html) - the GTDB RS226 entire database, and the NCBI Eukaryotic dindexed database.
Download the indexed GTDB RS226 database (Bacteria and Archaea). This requires 35 GB of disk space.
```
curl -L https://farm.cse.ucdavis.edu/~ctbrown/sourmash-db.new/gtdb-rs226/gtdb-rs226-k51.dna.rocksdb.tar.gz | tar xzf -
```
Download and unpack the Jan 2025 NCBI eukaryotes database. This requires 23 GB of disk space.
```
curl -L https://farm.cse.ucdavis.edu/~ctbrown/sourmash-db.new/ncbi-euks-2025.01/ncbi-euks-all-2025.01-k51.dna.rocksdb.tar.gz | tar xzf -
```
You should now have two directories, both ending in ``.rocksdb`:
```
ls -FC
```
>gtdb-rs226-k51.dna.rocksdb/
>ncbi-euks-all-2025.01-k51.dna.rocksdb/
## Download a metagenome to analyze
Grab a metagenome sig from our repository of all public SRA data sets:
```
curl -L https://wort.sourmash.bio/v1/view/sra/SRR11125891 -o SRR11125891.sig
```
You can replace `SRR11125891` with any public SRA accession you're interested in, [or sketch your own](https://sourmash.readthedocs.io/en/latest/sourmash-sketch.html).
## Run `sourmash gather` to select overlapping genomes
Now run sourmash gather to search the downloaded metagenome against both databases:
```
sourmash gather SRR11125891.sig \
gtdb-rs226-k51.dna.rocksdb \
ncbi-euks-all-2025.01-k51.dna.rocksdb \
-o SRR11125891.x.gtdb+euks.csv \
-k 51 --scaled 10_000
```
This will take ~1-2 min and require 1 GB of RAM.
The output of this command will be a CSV file that contains a [minimum metagenome cover](https://www.biorxiv.org/content/10.1101/2022.01.11.475838v2) listing a short list of known genomes that are present.
## Annotate the sourmash gather results with GTDB and NCBI eukaryote taxonomies.
Next we will generate a taxonomic summary based on the matching genomes. For this we will need to combine the GTDB taxonomy (for bacteria and archaea) with the NCBI eukaryotic taxonomy.
Start by downloading gtdb-rs226.lineages.csv and ncbi-eukaryotes.2025.01.lineages.csv, again, from the [prepared databases page](https://sourmash.readthedocs.io/en/latest/databases.html):
```
curl -JLO https://farm.cse.ucdavis.edu/~ctbrown/sourmash-db.new/gtdb-rs226/gtdb-rs226.lineages.csv
curl -JLO https://farm.cse.ucdavis.edu/~ctbrown/sourmash-db.new/ncbi-euks-2025.01/ncbi-eukaryotes.2025.01.lineages.csv
```
Then, use `sourmash tax` to annotate the gather output with taxonomy:
```
sourmash tax annotate -g SRR11125891.x.gtdb+euks.csv \
-t gtdb-rs226.lineages.csv \
ncbi-eukaryotes.2025.01.lineages.csv
```
Notes:
* you could also use the NCBI taxonomy instead of the GTDB taxonomy for the bacterial and archaeal matches, since every GTDB genome is also in NCBI.
* if you plan to run `sourmash tax` multiple times, you should use `sourmash tax prepare` to combine the CSV files into a SQLite database, which will be much faster!
## Create a taxburst taxonomy of the results
[taxburst](https://taxburst.github.io/) (a fork of [Krona](https://github.com/marbl/Krona)) is a nice way to explore taxonomic results.
Note the presence of some putative host contamination in this pig metagenome - the eukaryote 'Sus scrofa' is pig :).
Run:
```
taxburst -F tax_annotate SRR11125891.x.gtdb+euks.with-lineages.csv \
-o SRR11125891.x.gtdb+euks.taxburst.html
```
<iframe src="https://farm.cse.ucdavis.edu/~ctbrown/2025-euk-tax-tutorial-files/SRR11125891.x.gtdb+euks.taxburst.html" width="1200" height="800" frameborder="1" allowfullscreen></iframe>
(If anyone has better ideas for the iframe embedding, please let me know!)
## Generate a Sankey flow diagram of the results
Sankey flow diagrams are nice ways to view hierarchical decompositions! This is a nice interactive one, produced by [the betterplot plugin for sourmash](https://github.com/sourmash-bio/sourmash_plugin_betterplot/).
```
sourmash scripts sankey \
--annotate-csv SRR11125891.x.gtdb+euks.with-lineages.csv \
-o SRR11125891.x.gtdb+euks.sankey.html
```
<iframe src="https://farm.cse.ucdavis.edu/~ctbrown/2025-euk-tax-tutorial-files/SRR11125891.x.gtdb+euks.sankey.html" width="1200" height="800" frameborder="1" allowfullscreen></iframe>
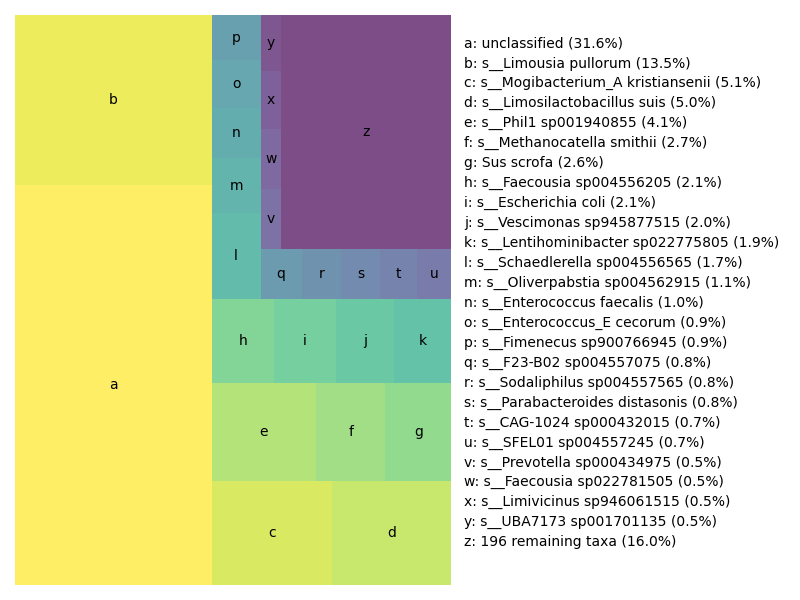
## Generate a treemap of the results
Last but not least, here is a proportional representation of species detected in the metagenome. This is a 'treemap', which does a nice job of showing the content weighting.
First we need a new format for the taxonomic summary:
```
sourmash tax metagenome -F csv_summary \
-o SRR11125891.x.gtdb+euks \
-g SRR11125891.x.gtdb+euks.csv \
-t gtdb-rs226.lineages.csv \
ncbi-eukaryotes.2025.01.lineages.csv
```
and then we run treemap (again from [the betterplot plugin for sourmash](https://github.com/sourmash-bio/sourmash_plugin_betterplot/):
```
sourmash scripts treemap \
SRR11125891.x.gtdb+euks.summarized.csv \
-o SRR11125891.x.gtdb+euks.treemap.png \
-r species
```
The file `SRR11125891.x.gtdb+euks.treemap.png` will contain the following image:
## Additional notes and discussion points
### Why are we using k=51?
K=51 is a very stringent match, and we are certainly missing some bacterial content here. However, we've found that lower k-mer sizes introduce too many false positives with large eukaryotic genomes. We're working on it!
### Can we do a simultaneous search for viruses?
We do have viral databases [for sourmash](https://sourmash.readthedocs.io/en/latest/databases.html), but the search parameters are incompatible with k=51/scaled=10_000. We're working on it :). See [sourmash-bio/sourmash#1340](https://github.com/sourmash-bio/sourmash/issues/1340).







