---
tags: SIO, workshop, 2017
---
# [Course Post Survey](https://docs.google.com/forms/d/e/1FAIpQLSfTYX0kT3okTfU0p8n96N0oLD_D3sY1LxPPHdCPUUm1BDDnKA/viewform)
SIO Transcriptomics Workshop: Thursday collaborative notes
============
#### October 12, 2017
### **[Course website](http://rnaseq-workshop-2017.readthedocs.io/en/latest/index.html)**
#### Twitter hashtag: [#SIOrnaseq2017](https://twitter.com/search?q=%23SIOrnaseq2017&src=typd)
https://twitter.com/search
---
### **Signin for 10/12**
1. John McCrow (JCVI)
2. Faeazeh Ghaz(UCSD, BO)
3. Alice Harada (SIO, MB)
4. Stephanie Sommer (SIO, BO)
5. Henrique Machado (SIO, CMBB)
6. Kaitlin Creamer (SIO, CMBB)
7. Doug Sweeney (SIO, CMBB MB)
8. Grant Batzel (SIO, MBRD)
9. Catherine Schrankel (SIO, MBRD)
10. Sara R. Rivera (SIO, MCG)
11. Yaqun Zhang (SIO,JCVI)
12. Sarah Schwenck (SIO, BO)
13. Charlotte Seid (SIO, BIC)
14. Julie Dinasquet (SIO, MBRD)
15. Lauren Manck (SIO, MCG)
16. Lisa Komoroske (NOAA, UMass)
17. Deirdre Lyons (SIO, MBRD)
18. Ryan Guillemette (SIO, MBRD)
19. Tessa Pierce (UC Davis)
20. Lisa Johnson (UC Davis)
21. Natalya Gallo (SIO BO)
22. Evelien De Meulenaere (SIO, MBRD)
23. Du Niu (JCVI/SIO)
24. Jessica Blanton (SIO,MBRD)
### Questions from the audience:
(continued from Wednesday)
1. Who are the local researchers working heavily in RNAseq Analysis
2. What other local training resources are available (workshop, bootcamp, dept. courses) about RNA-seq data and gene expression data analyses?
- Terry's class (on main campus?)-can someone add the course # here who knows?
- Also we encourage folks to not just use local resources-very active online community (forums, twitter, etc.) that are great resources, especially for up and coming techniques/programs
- PAG is in San Diego every Jan -if can't go, can follow on twitter www.intlpag.org/
3. DN: I am going to an illumina workshop today from 11-1:30, let me know if you have questions for their sequence team
- curious about their take on MACE as alternative to RNA-Seq http://genxpro.net/sequencing/transcriptome/mace-massive-analysis-of-cdna-ends/
- It's a tough question, and the answer to it depends on the budget and what type of questions to be asked. Usually MACE is something to start with for the budget and convenience, however with the cost for deeper sequencing going down, "I" think the full mRNA Seq would be preferred even for the early stage of a study.
4. When confirming with Sanger sequencing, which to trust more - Illumina assembly or Sanger base call?
- Illumina seq yields more depth (repetitive reads of the same sequence) than Sanger seq. If majority of Illumina seq reads have certain base at position of interest vs. Sanger seq, would trust Illumina more. See [Summary of basecalling on NGS](https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3178052/) and [Tech note on Illumina](https://www.illumina.com/documents/products/technotes/technote_Q-Scores.pdf)
5. Can you go over how computing time works?
- For XSEDE, service units (SUs) are calculated by the number of hours you need to use (how long the programs take to run) multiplied by the number of CPU you need to run each of the programs. "Wall time" means how long it actually takes to run. "CPU time" is the amount of time the computer is being occupied, which is more than wall time because sometimes processes can be run in parallel on multiple processors.
6. Will there be time to quickly discuss how best to present/make figures/etc from transcriptomics data? Tools, best practices, suggestions, to do/don't do, etc.
- While this depends on your type of data and questions, there are some common ones that we'll go over in the DE modules and visualization using Juypter notebooks. We can also add links below for more resources...
7. experimental design: biological vs technical replicates, library prep methods and reagents (NEB vs Illumina kits), resources at SIO for checking sample quality (e.g. TAPE station?), and where to send samples...UCSD or elsewhere?
- UCSD genome core http://igm.ucsd.edu/genomics/services.shtml
- TSRI https://www.scripps.edu/california/research/ngs/index.html
- For library QC (bioanalyzer, qubit, tape station), the genome core facilities can do this for you for a cost; since we (the instructors) aren't at SIO, we don't know which labs have this equipment-would suggest talking to the labs at SIO that do NGS and ask what they use...(and/or if anyone in the class is in a lab that has their own please edit this)
- Biological replicates are much more important than technical replicates, especially with barcode/multiplexing that is now common (i.e., lane/batch effects are real, but avoided by multiplexing across lanes). We can add some papers/tech notes about this below...
- Library prep methods-there are some common ones but again depends on your sample type, etc.
- Whitehead lab uses modified NEB, protocols here:
https://github.com/WhiteheadLab/Bench_Protocols
- Can others please add info on the library prep methods they use/suggest/not like here...?
-
**8. How to choose which tool for each step and where to get info of the best fit for your goal?**
9. Similar to the previous question, but for sample preparation:
? what are the different steps? => sample storage (freezing/RNAlater/...), lysis+extraction, fragmentation?
? anyone have an opinion on better kits for different parts of the preparation?
? RNA isolation from bacteria is pretty straightforward but from invertebrates (no blood..) seems to have a rather limited amount of choices to use.
? How do you check your sample/library quality?
-
### Metatranscriptomics: have you ever compared orthomcl to proteinortho? how do they compare?**
- OrthoMCL is somewhat difficult to install, but the MCL part is not, which takes a allvall BLAST
- tSNE clustering - iterative markov model
Quantification:
Why Quantify? (insert tessa's slide)
### Experimental Design Discussion
*What factors influence study design?*
- Cost
- Replication is essential
- Balancing cost with number of samples/conditions and replicates
- How many replicates are required? At least 3, but need to consider how much variation you expect
- Consider ahead of time how you will analyze the data when planning how to run the experiment or sampling
- Could do a small test before conducting the full study
- Randomize as much as possible
*How do I know if I will have enough power to answer my question?*
- Statistical power is a measure of how likely you are to be able to detect differences between a treatment and control, for example, given the amount of difference in expression values, and variation.
- Consider effect size, estimate of variation, and sample size
*What are potential challenges and limitations?*
- Sequencing can give insight, but does not equate to biological understanding
- Sample quality, quantity of RNA
- Analysis requires a lot of time and a variety of skillset
- The transcriptome is not everything. Consider post-translational modifications and regulation. Might want to integrate other sources of information, genomics, proteomics, etc.
- De Novo Transcriptomes- how to identify **contamination** if you do not have a reference
- Homology search vs database (e.g. NCBI nr) may give highlight what organisms your transcripts belong to.
- [DarkHorse software](http://darkhorse.ucsd.edu/download.html) (Podell et al, Eric Allen's group) is a great program to associate phylogenetic linages with your sequences. Usefull in Genomics, Transcriptomics, shotgun metagenomic data.
- SIO-BUG November meeting will probably touch on this
### Analysis info and Resources Discussion (in addition to those listed on workshop website)
Papers:
- [A survey of best practices for RNA-seq data analysis](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8)
- [Voom: precision weights unlock linear model analysis tools for RNA-seq read counts](https://genomebiology.biomedcentral.com/articles/10.1186/gb-2014-15-2-r29)
- [Deep Evolutionary Comparison of Gene Expression Identifies Parallel Recruitment of Trans-Factors in Two Independent Origins of C4 Photosynthesis](http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004365)
Programs/Packages
(Note: there are a TON and many are best for different types of data/situations; we are listing here just a smattering of ones we use/see commonly but by no means are they always best, etc)
- [Tabview](https://.com/TabViewer/tabview). Install: `sudo pip install tabview`
- [Pandoc](https://pandoc.org/) a universal document converter
- [Limma tutorial with filter plot code](https://www.bioconductor.org/help/workflows/RNAseq123/)
- Online resources (tutorials & online forums, etc)
- https://www.biostars.org/
- http://seqanswers.com/forums/
- https://support.bioconductor.org/
### Resources for applying for Jetstream/XSEDE allocation
- Submit via User portal: https://portal.xsede.org/submit-request#/
- Successful applications:
- [Collection](https://github.com/ljcohen/jetstream-xsede-illo/tree/master/xsede_applications)
- [this course](https://github.com/bluegenes/proposals/blob/master/xsede/RNAseq_workshop_Oct2017.md)
- [from XSEDE](https://portal.xsede.org/allocations/startup#examples)
- [Titus Brown for DIBSI at UCDavis](http://ivory.idyll.org/blog/2017-dibsi-xsede-request.html)
- [Requirements for application](https://portal.xsede.org/allocations/startup#requiredcomponents)
### How to Set Up Genomics Workflows on the Amazon Web Services (AWS) Cloud
Date: Tuesday October 24th, 2017
Time: 1:30pm – 3:30pm
Location: UCSD – Goldberg Auditorium Room 2110
[Register Here](http://www.awspartnernet.apncampaigns.com/sw/swchannel/Registration/internet/Registration.cfm?SWSESSIONID=4E18C0924E5495BD7B40436CF7B51872&RegPageID=3063796)
### Sign up for Github!
https://github.com/
Helps you keep track of your own scripts and files, organized by project. For easy collaborations with colleagues - and your future self!
* [Interactive lesson](http://rnaseq-workshop-2017.readthedocs.io/en/latest/LC-github.html)
* Github organization account for [Open Data Science at SIO](https://github.com/Open-Data-Science-at-SIO)
---
---
SIO Transcriptomics Workshop: Wednesday collaborative notes
============
#### October 11, 2017
### **[Course website](http://rnaseq-workshop-2017.readthedocs.io/en/latest/index.html)**
### Twitter hashtag: [#SIOrnaseq2017](https://twitter.com/search?q=%23SIOrnaseq2017&src=typd)
**Course Mechanics**
- Ask questions!
- Use stickies (put up red for "help", put up blue for "all-good")
To-do list:
* [x] Intro slides
* [x] Boot your jetstream instance
### Discussion: Intro to RNAseq
(take some notes here)
---
### **Signin for 10/11**
1. Lisa Johnson (UC Davis)
1. John McCrow (JCVI)
1. Tessa Pierce (UC Davis)
1. Lisa Komoroske (NOAA, UMass)
1. Jessica Blanton, (SIO, MBRD)
1. Sara R. Rivera (SIO, MCG)
1. Yaqun Zhang (JCVI, SIO)
1. Du Niu (JCVI/SIO)
1. Natalya Gallo (SIO, BO)
1. Charlotte Seid (SIO, BIC)
1. Sarah Schwenck (SIO, BO)
1. Stephanie Sommer (SIO, BO)
1. Evelien De Meulenaere (SIO, MBRD)
1. Ryan Guillemette (SIO, MBRD)
1. Grant Batzel (SIO, MB)
1. Doug Sweeney (SIO, MB)
1. Alice Harada (SIO, MBRD)
1. Ariel Rabines, (SIO/JCVI)
1. Julie Dinasquet (SIO, MBRD)
1. Kaitlin Creamer (SIO, CMBB)
1. Henrique Machado (SIO, CMBB)
1. Angela Zoumplis (SIO, MB)
1. Roshan Shrestha (SIO)
1. Lauren Manck (SIO, MCG)
1. Dede Lyons (SIO/MBRD/H.sapiens)
1. Catherine Schrankel (SIO/MBRD)
1. Phillip Morin (SWFSC)
1. Tracey Somera (SIO, CMBB)
1. Mike Tift (SIO, CMBB)
1. Tiago Leao (SIO, CMBB)
---
### Morning Session discussion
**Prompt1**: What is Transcriptomics?
**Prompt2**: When would Transcriptomics be a good tool?
- Core functional expression analyses
- NOT best for ribosomal analyses
- Population genetics is also a use of the same data (doeNOT expression work)
- Anything seen in RNAseq data is best complemented by alternative validation (qPCR, protein)
- Working with non model organisms- there are techniques to get around not having a genome. This certainly a current topic in transcriptomics methods
---
### Jetstream cloud computing rosetta stone
- "Instance" = computer
- "Volume" = harddrive
Each instance on Jetstream is like a new computer. You must make sure that you install the programs and/or languages that you want to use before you can run any analyses. You will also need to transfer your data from your local machine or other remote server to a folder on your instance on Jetstream.
When you close an instance on Jetstream, you will lose all the files you have saved on it, as well as any programs you have installed.
**What does the data look like?**
Illumina paired reads are short fragments which cover regions of the DNA you are sequencing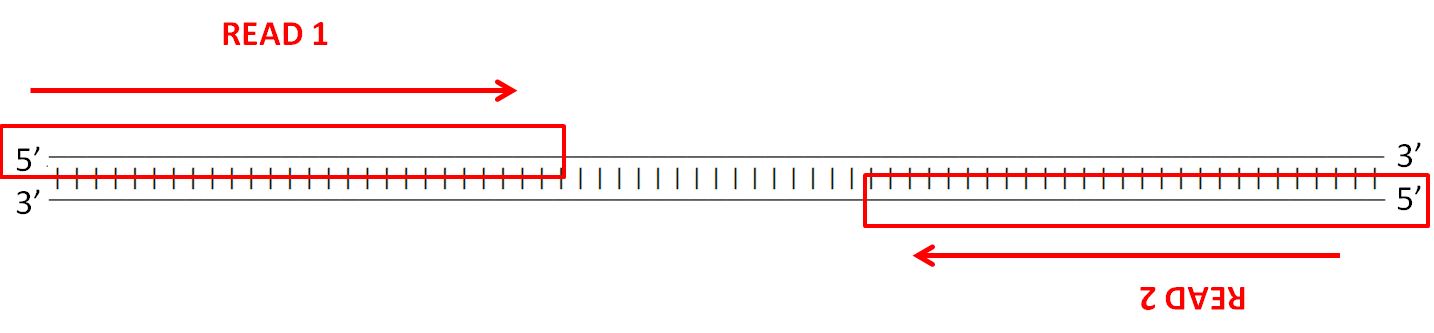
[**Video of illumina sequencing by synthesis](https://www.youtube.com/watch?v=fCd6B5HRaZ8)
___
### Questions from the audience
**After the course can we use the computing hours leftover for our own project?** ~~*(In short, no. But we will/can discuss your options!)*~~ **YES!**
we will discuss Thursday - https://www.xsede.org/
1. Where do I store all of my data?
- Private: hardrive,
- Private Cloud: OneDrive, unlimited Google through UCSD (ask IT!!)
- UCSD library
- Public: [Sequence Read Archive (NCBI SRA)](https://www.ncbi.nlm.nih.gov/sra), See [submission guidelines](https://submit.ncbi.nlm.nih.gov/subs/sra/)
- The UCSD Library offers free data storage service. One is a staging server space. Contact Ho Jung Yoo <hjsyoo@ucsd.edu> and Tim Marconi <tmarconi@ucsd.edu> to discuss your data storage needs and they will get you set up. Possible to store hundreds of TB of data (and also send large amounts of data to others). All stored privately.
2. How much do I need to know about my sample in order to gain meaningful data from RNA-seq? e.g. for model bacteria, must one have a well annotated genome? what happens if you do not?
- We will address further Thursday morning in our experimental design discussion
3. Can we discuss clustering methods to identify genes that may be co-regulated/co-expressed?
- Yes -John will include on Thursday an example from OrthoMCL-we can add more discussion of other options if there is interest http://orthomcl.org/orthomcl/
4. Dealing with multigene families: Issues in mapping and understanding correct signal vs mapping errors
5. Is it possible to recover RNA-seq data for chl and mito genes if poly-A method was used?
- Yes. It is possible, there is usually some low level of sequences that were not poly-A selected but still remain in the sample
6. What happens to the files and analysis done on jetstream once your allocated time is over?
7. SIO resources for library prep design, generating RNA-seq libraries and storing data: Kit recommendations etc
8. Resources to understand how to spike experiments?
9. How to choose the proper sequencer for your project? e.g. 50 pair reads but less samples per line, or 140 pairs reads with more samples per line. Any suggestions?
10. Naming of the files and keeping track - best practices and ideas?!
- Don't use spaces in file names, use "_" instead.
- Keep relevant data in the name, such as sample, date, condition, etc.
- Be consistent and rename files to your own style so they are easy to understand and organize
- Keep a metadata file with keys explaining what any shorthand means.
11. Alternative to Trimmomatic = BBtools. https://jgi.doe.gov/data-and-tools/bbtools/
12. If your mRNA sequences are hitting intron regions in a reference genome, does this suggest the annotation is wrong? Or is this just alternative splicing?
13. How many replicates are necessary / practical? What type of statistics are people using for their data?
14. Can you give us an idea of costs associated with transcriptomics?
---
### Conda for package management and virtual environments
Conda -- which can be downloaded as Miniconda or Anaconda -- is a Python distribution, package manager, and virtual environment solution. For a lightweight installation of Conda, install [Miniconda](http://conda.pydata.org/miniconda.html) (recommended: use the Python 3 version).
To use Conda, you can create a virtual environment and install some software into it. For example, to set up a basic data science environment, you can run this command:
conda create -n myenv jupyter pandas seaborn
Then activate your environment:
source activate myenv
Then do some work! For example, open a Jupyter Notebook by typing `jupyter`.
When you are done working, deactivate the environment:
source deactivate myenv
---
### Random
* [xkcd: tar](https://xkcd.com/1168/)
* [xkcd: Sandwich](https://xkcd.com/149/)







