# Sequence-a-Genome 2023
Jason - willliams@cshl.edu
Anna - feitzin@cshl.edu
---
### Learning Resources
- CyVerse [link](https://learning.cyverse.org)
- Genomics data carpentry: https://datacarpentry.org/lessons/#genomics-workshop
- [Shell lesson](https://datacarpentry.org/shell-genomics/)
**General Coding**
- CodeCademy: [link](https://www.codecademy.com/)
- Hour of code (also in languages other than English): [link](https://code.org/learn)
**Software installations**
Be sure you have permission to install software
- Try Ubuntu: [link](https://tutorials.ubuntu.com/tutorial/try-ubuntu-before-you-install#0)
- Python: [link](https://www.python.org/dowloads/)
- Jupyter: [link](https://jupyter.org/)
- Wing IDE: [link](https://wingware.com/)
- Atom text editor: [link](https://atom.io/)
**Bioinformatics**
- Learn bioinformatics in 100 hours: [link](https://www.biostarhandbook.com/edu/course/1/)
- Rosalind bioinformatics: [link](http://rosalind.info/about/)
- Bioinformatics coursera: [link](https://www.coursera.org/learn/bioinformatics)
- Bioinformatics careers: [link](https://www.iscb.org/bioinformatics-resources-for-high-schools/careers-in-bioinformatics)
**Help**
- General software help: [link](https://stackoverflow.com/)
- Bioinformatics-specific software help: [link](https://www.biostars.org/)
### DNAi Videos
- Sequencing project animation [link](https://youtu.be/-gVh3z6MwdU)
- Beginnings of the Human Genome Project at the Cold Spring Harbor Laboratory, James Watson [link](https://dnalc.cshl.edu/view/15445-Beginnings-of-the-Human-Genome-Project-at-the-Cold-Spring-Harbor-Laboratory-James-Watson.html)
- Importance of genetic maps, Mary-Claire King [link](https://dnalc.cshl.edu/view/15128-Importance-of-genetic-maps-Mary-Claire-King.html)
- Compiling the data from the Human Genome Project, Jim Kent [link](https://dnalc.cshl.edu/view/15305-Compiling-the-data-from-the-Human-Genome-Project-Jim-Kent.html)
- Using computers to predict how genes within the human genome, Craig Venter [link](https://dnalc.cshl.edu/view/15358-Using-computers-to-predict-how-genes-within-the-human-genome-Craig-Venter.html)
- Finding genes in the human genome, Ewan Birney [link](https://dnalc.cshl.edu/view/15291-Finding-genes-in-the-human-genome-Ewan-Birney.html)
- The public Human Genome Project's DNA donors, Eric Lander [link](https://dnalc.cshl.edu/view/15327-The-public-Human-Genome-Project-s-DNA-donors-Eric-Lander.html)
- The first draft of the human genome, Ari Patrinos [link](https://dnalc.cshl.edu/view/15343-The-first-draft-of-the-human-genome-Ari-Patrinos.html)
- Relating a gene to a sequence of amino acids, Sydney Brenner [link](https://dnalc.cshl.edu/view/15279-Relating-a-gene-to-a-sequence-of-amino-acids-Sydney-Brenner.html)
---
### Other Important Links
- Human genome at NCBI: [link](https://www.ncbi.nlm.nih.gov/genome/guide/human/)
- Markdown cheatsheet: [link](https://github.com/adam-p/markdown-here/wiki/Markdown-Cheatsheet)
- AllofUs: https://allofus.nih.gov/
### Laboratory
- Plant DNA extraction: [link](https://www.promega.com/products/nucleic-acid-extraction/genomic-dna/high-molecular-weight-dna-extraction-kit/?catNum=A2920#protocols)
- Microbial swab DNA extraction: [llink](https://jasonjwilliamsny.github.io/stars-2022/documentation/microbiome_dna_isolation/)
### Software
**Software Utilities**
- PuTTY for windows: https://the.earth.li/~sgtatham/putty/latest/w64/putty.exe
- Install Docker on Ubuntu: https://docs.docker.com/engine/install/ubuntu/
- Miniconda: https://docs.conda.io/en/latest/miniconda.html#linux-installers
- Bioconda: https://bioconda.github.io/user/install.html
- Samtools manual: http://www.htslib.org/doc/
- IGV: https://software.broadinstitute.org/software/igv/
- Filezilla (client): https://filezilla-project.org/
---
## Monday
- [x] Introduction
- [x] Microbial DNA Isolation [protocol](https://jasonjwilliamsny.github.io/stars-2022/documentation/microbiome_dna_isolation/)
- [x] Spinach Chloroplast Isolation [protocol](https://cdn.shopify.com/s/files/1/0063/4575/2687/files/11-CP-11_v6.pdf?v=1613417694
- [x] SeekApp by iNaturalist [App link](https://www.inaturalist.org/pages/seek_app
- [x] Database of sequenced chloroplast genomes [link](https://www.gndu.ac.in/CpGDB/ChloroplastGenomeInformationRetrievalSystem.aspx)
#### Shoe swab samples
|Samples (A=Norgen, B=Chelex)|Barcodes (A,B)|Name|
|----------------------------|--------------|----|
|1a,1b|1,2|Kevin|
|2a,2b|3,4|David|
|3a,3b|5,6|Peter|
|4a,4b|7,8|Lucas|
|5a,5b|9,10|Kamryn|
|6a,6b|11,12|Rishi|
|7a,7b|13,14|Aine|
|8a,8b|15,16|Jupiter|
|9a,9b|17,18|Jef|
|10a,10b|19,20|Daniel|
|11a,11b|21,22|Keith|
|12a,12b|23,24|Anna|
Worked: 1,3,5,8,14,15,16,19,23
## Tuesday
- [x] Chloroplast isolation from student samples
- [x] Prepare 2 preps per plant
- [x] Elute both samples in 50ul cold PBS and combine
- [x] Monarch DNA extraction from isolated chloroplasts
- [x] [Chloroplast isolation protocol](https://cdn.shopify.com/s/files/1/0063/4575/2687/files/11-CP-11_v6.pdf?v=1613417694
- **MODIFY LAST STEP - elute each prep in 50ul of PBS and combine **
- [x] [NEB Monarch kits - cell culture protocol](https://www.neb.com/protocols/2018/10/23/protocol-for-extraction-and-purification-of-genomic-dna-from-cells-t3010)
- [x] Gel electrophoresis of 16s
- [x] History of DNA sequencing
### Monarch DNA extraction flow chart
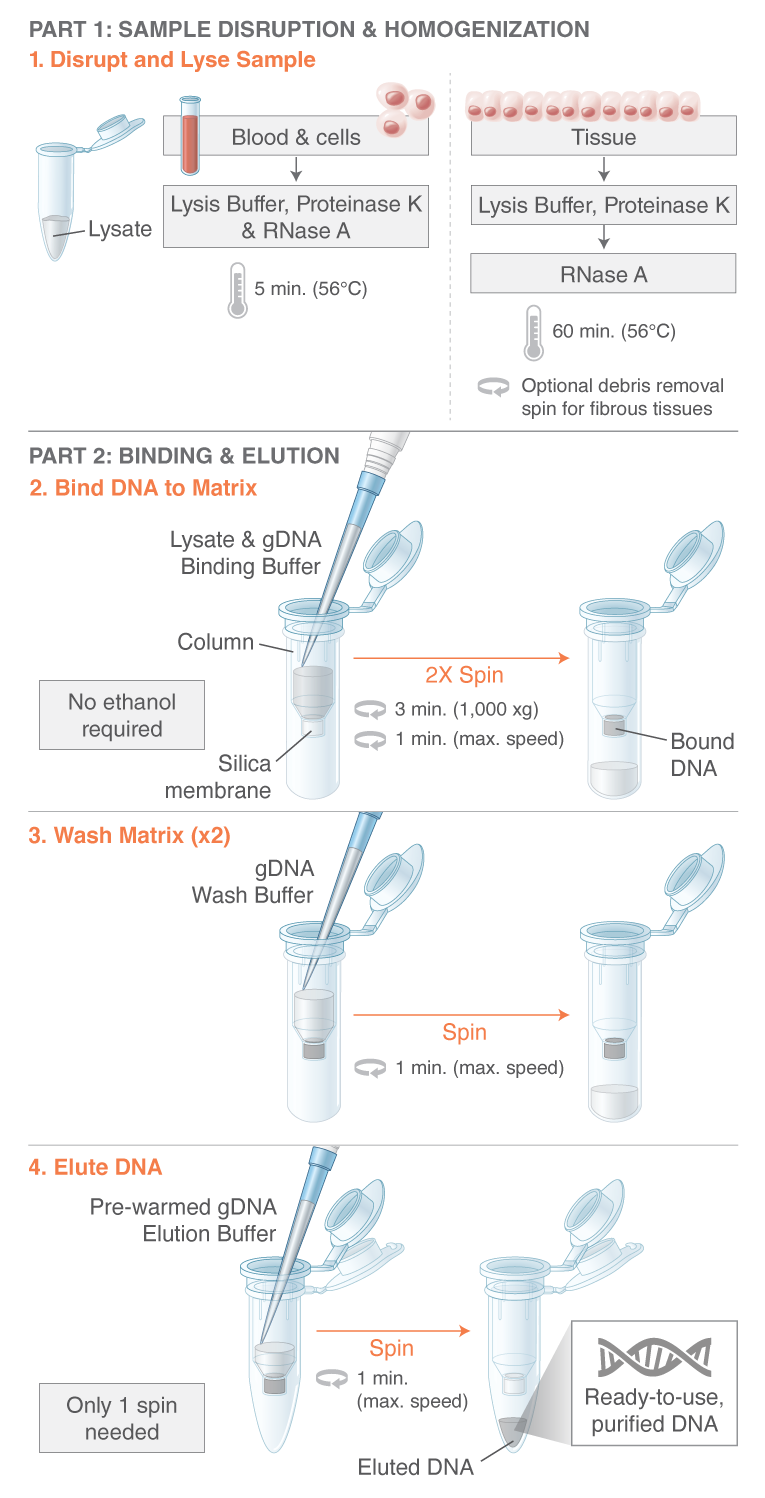
## Student samples
**Place photos of your plant in our [Google Drive](https://drive.google.com/drive/folders/1DsIPPgNhceZE2u0f5x8UoamLqab_sefC?usp=sharing)**
- Ensure photos are labeled with your number
|Sample|Tentative identification|Nanopore barcode|Name|Notes|Concentration (ng/ul)|Low input?|Reference genome|Genome size|Num Contigs|Largest Frag|
|------|------------------------|----------------|----|-----|---------------------|----------|----------------|-----------|-----------|--------------|
|1|Japanese Stiltgrass|80|Kevin|Chloroplasts|1.24|Y|[Andropogon burmanicus](https://www.ncbi.nlm.nih.gov/nuccore/NC_035038)|140898 bp|55|19058|
|2|Dicot/Flower Plant|79|David|Chloroplasts|4.87|Y|[Acer pseudosieboldianum](https://www.ncbi.nlm.nih.gov/nucleotide/NC_046487.1)|157053 bp|57|21874|
|3|Fuji Apple Tree(Malus pumila)|78|Peter|Whole Tissue|23.4||[Malus micromalus](https://www.ncbi.nlm.nih.gov/nuccore/NC_036368)|159834|120|53221|
|4|Pieris japonica (Japanese andromeda)|77|Lucas|Whole tissue|27.0||[Vaccinium oldhamii](https://www.ncbi.nlm.nih.gov/nuccore/NC_042713)|173245bp|148|58789 |
|5|Cherry laurel|76|Kamryn|Chloroplasts|4.25|Y|[Prunus serotina](https://www.ncbi.nlm.nih.gov/nuccore/NC_036133)|158778 bp|41|14449|
|6|Hydrangea|75|Rishi|Whole tissue|23.0||[Hydrangea paniculata](https://www.ncbi.nlm.nih.gov/nuccore/NC_044829)|157881 bp|||
|7|Red Maple|74|Aine|Chloroplasts|6.20|Y|[Acer miaotaiense](https://www.ncbi.nlm.nih.gov/nuccore/NC_030343)|156595 bp|15|29337|
|8|Begonia|73|Jupiter|chloroplasts|2.97|Y|[Begonia pulchrifolia](https://www.ncbi.nlm.nih.gov/nuccore/NC_045096)|169589 bp|16|76127|
|9|Rosa Chinesis|72|Jef|Whole tissue|3.96|Y|[Rosa chinensis](https://www.ncbi.nlm.nih.gov/nuccore/NC_038102)|156590 bp|222|190771|
|10|Perilla frutescens|71|Daniel|Chloroplasts|4.62|Y|[Perilla frutescens](https://www.ncbi.nlm.nih.gov/nuccore/NC_030755)|152588 bp|64|83591|
|11|Polemoniaceae(Phlox)|70|Anna|Chloroplast|11.3|Y|[Polemonium chinense chloroplast](https://www.ncbi.nlm.nih.gov/nuccore/MN057953.1)|155578 bp |79|107753|
|12|Pumpkin (Long Island Cheese)|69|Jason|Chloroplasts|26.1||[Cucurbita moschata](https://www.ncbi.nlm.nih.gov/nuccore/NC_036506)|157644 bp |220|190771|
|13|Boxwood|68|Keith|Chloroplasts|0.893|Y|[Buxus microphylla](https://www.ncbi.nlm.nih.gov/nuccore/NC_009599)|159010 bp|17|23428
#### Low input barcode ligation recipie
|Reagent|Amount|
|-------|------|
|End-prepped DNA|4µl|
|Native barcode |1.25 µl|
|Blunt/TA Ligase Master Mix |5 µl|
|Total|10.25 µl|
**For "high" input 1ul of DNA is OK**
## Wednesday
- [x] Quantification of chloroplast DNA preps
- [x] [Qubit protocol](https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2FMAN0017455_Qubit_1X_dsDNA_HS_Assay_Kit_UG.pdf)
- [x] Prep of 16s experiment libary
- [x] Introduction to Nanopore
- [x] Sequencing on Nanopore
### PCR Cleanup for 16s
1. Transfer each sample to a separate 1.5 ml DNA LoBind Eppendorf tube.
2. Add 30 µl of resuspended AMPure XP beads to the reaction and mix by pipetting.
3. Incubate on a Hula mixer (rotator mixer) for 5 minutes at room temperature.
4. Prepare 500 μl of fresh 70% ethanol in nuclease-free water.
5. Spin down the sample and pellet on a magnet. Keep the tube on the magnet, and pipette off the supernatant.Keep the tube on the magnet and wash the beads with 200 µl of freshly prepared 70% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
6. Repeat the previous step.
7. Spin down and place the tube back on the magnet. Pipette off any residual ethanol. Allow to dry for ~30 seconds, but do not dry the pellet to the point of cracking.
8. Remove the tube from the magnetic rack and resuspend pellet in 10 µl of 10 mM Tris-HCl pH 8.0 with 50 mM NaCl. Incubate for 2 minutes at room temperature.
9. Pellet the beads on a magnet until the eluate is clear and colourless.
10. Remove and retain 10 µl of eluate into a clean 1.5 ml Eppendorf DNA LoBind tube.
### Native barcoding of chloroplast DNA
#### Workflow

## Thursday
```Note
#merge fastq files
cat FAS*.fastq.gz >> merged_fastq.fasq
# map to reference with minimap2
minimap2 -t 14 -x map-ont -a ~/workdir/mappings/basecall_tiny_porechopped_vs_wuhan.sam ~/workdir/wuhan.fasta ~/workdir/data_artic/basecall_tiny_porechopped.fastq.gz
SAM to FastQ
grep -v ^@ INPUT.sam | awk '{print "@"$1"\n"$10"\n+\n"$11}' > OUTPUT.fastq
## install flye
sudo conda create -y --name flye flye==2.9.2 -c bioconda
## assemble genome with flye
flye --nano-raw OUTPUT.fastq -o assembly
```
#### Sequencing results
**16s Sequencing results**
- Run time: 2 hr 55 min
- Read count: 744 k
- Basecalled: 1.09 Gb
- [Epi2Me Report](https://epi2me.nanoporetech.com/shared-report-421789?tokenv2=f1e99fa2-a4fe-45b6-8eab-c0eef7717b24)
**Chloroplast sequencing results**
- Run time: 16 hrs 3 min
- Read count: 2.02 M
- Basecalled: 2.34 Gb
####
IP Information
- Kevin 149.165.152.183
- David 149.165.159.206
- Peter 149.165.159.186
- Lucas 149.165.159.213
- Name 149.165.159.214
- Rishi 149.165.159.15
- Kamryn 149.165.159.107
- Jupiter 149.165.159.76
- Jef 149.165.152.86
- Daniel 149.165.159.94
- Aine 149.165.159.143
- Anna 149.165.159.153
- Jason 149.165.152.250
- Keith 149.165.159.82
### Software installations
```
# Install fastp with bioconda
sudo conda create -y --name fastp fastp==0.20.0 -c bioconda
```
```
# Create a shortcut to some example reads
ln -s /mnt/ceph/tutorial_example/fastp_results/1000_reads ~/example_reads
# copy some example fastq reads to a dir called test_fastp
cp example_reads/1000_called_reads_filtered.fastq.gz test_fastp/
#look at the first four lines of the compressed fastq file
zcat 1000_called_reads_filtered.fastq.gz |head -n4
```
```
# initialize conda in shell
conda init bash
# close terminal and reopen; load bash
bash
# activate the fastp enviornment
conda activate fastp
# move to directory with reads
cd test_fastp/
# run fastp to filter reads less than 2000
fastp -l 2000 -i 1000_called_reads_filtered.fastq.gz -o 1000_called_reads_filtered_2000.fastq.gz
```
## Friday
```
# Copy the fastq reads to our raw_fastq folder
cp /mnt/ceph/sag-2023/seq-23-chloroplast/fastq_pass/barcodeXX/*.fastq.gz ~/Documents/sag_2023/cp_experiment/raw_fastq/
# Confirm that the copying command worked
ls ~/Documents/sag_2023/cp_experiment/raw_fastq/
# Change into the raw_fastq dir
cd ~/Documents/sag_2023/cp_experiment/raw_fastq/
# Concatenate the reads into one file
cat *.fastq.gz >> merged_XX.fastq.gz
# make a dir for filtered reads
mkdir ~/Documents/sag_2023/cp_experiment/filtered_fastq
# make sure fastp is activated
conda activate fastp
# Run fastp MUST ALTER THE COMMAND TO YOUR NUMBER
fastp -i ~/Documents/sag_2023/cp_experiment/raw_fastq/merged_XX.fastq.gz -o ~/Documents/sag_2023/cp_experiment/filtered_fastq/filtered_merged_XX.fastq.gz -l 1000
# Create a porechop enviornment
sudo conda create -y --name porechop porechop_abi -c bioconda -c conda-forge
# activate porechop tool
conda activate porechop
#Run porechop (removes barcodes and adapters before assembly)
porechop_abi --ab_initio -i ~/Documents/sag_2023/cp_experiment/filtered_fastq/filtered_merged_XX.fastq.gz -o ~/Documents/sag_2023/cp_experiment/filtered_fastq/chopped_filtered_merged_XX.fastq.gz
# Create a dir for your mapping
mkdir ~/Documents/sag_2023/cp_experiment/mapped_reads
cd ~/Documents/sag_2023/cp_experiment/mapped_reads
# Create a minimap2 enviornment minimap2
sudo conda create -y --name minimap2 minimap2 -c bioconda
# activate minimap2
conda activate minimap2
# Remind yourself of your ref genome file name
ls ~/Documents/sag_2023/cp_experiment/ref_genome/
# Map your reads to a reference genome
minimap2 -t 8 -ax map-ont ~/Documents/sag_2023/cp_experiment/ref_genome/XXXXXXXXXXX_genome.fasta ~/Documents/sag_2023/cp_experiment/filtered_fastq/chopped_filtered_merged_XX.fastq.gz > ~/Documents/sag_2023/cp_experiment/mapped_reads/alignment.sam
# Check you are in the correct directory
~/Documents/sag_2023/cp_experiment/mapped_reads
# Extract aligned reads from sam mapping
grep -v ^@ alignment.sam | awk '{print "@"$1"\n"$10"\n+\n"$11}' > chloroplast_reads.fastq
# Create a new dir for assembly
mkdir ~/Documents/sag_2023/cp_experiment/flye_assembly
# Install flye
sudo conda create -y --name flye flye -c bioconda
# Activate flye
conda activate flye
# Change to the flye dir
cd ~/Documents/sag_2023/cp_experiment/flye_assembly
# Run flye
flye --nano-raw ~/Documents/sag_2023/cp_experiment/mapped_reads/chloroplast_reads.fastq -o ~/Documents/sag_2023/cp_experiment/flye_assembly/
#fixing a cp file
(@.*\n)\*\n\+\n\*
```
### Recovering from an error
```
# Install Samtools
sudo conda create -y --name samtools samtools -c bioconda
# Install missing library
# Say Yes (Y) when asked to install
sudo apt-get install libncurses5
# Go back to the dir with the mapped alignment
cd ~/Documents/sag_2023/cp_experiment/mapped_reads
# Activate same tools and create a new fastq file
conda activate samtools
samtools fastq alignment.sam >sam_out.fastq
# Go back to the flye assembly directory
cd ~/Documents/sag_2023/cp_experiment/flye_assembly
# activate flye and run
conda activate flye
flye --nano-raw ~/Documents/sag_2023/cp_experiment/mapped_reads/sam_out.fastq -o ~/Documents/sag_2023/cp_experiment/flye_assembly/
```
### Annotate your genome
[Online chloroplast annotation tool](https://chlorobox.mpimp-golm.mpg.de/geseq.html#)
 Sign in with Wallet
Sign in with Wallet







