###### tags: `Linux` `Unix` `Shell` `Bash` `awk` `grep` `for` `sort` `tail` `cat` `tail` `sed` `tr` `paste` `regex` `uniq` `ls` `realpath` `wc` `echo` `cut` `which` `cp` `scp` `find` `exec` `mv` `rm` `ls` `realpath` `xargs` `basename` `dirname` `dos2unix` `od` `EOL` `notepad++` `windows characters` `od` `mkdir` `crontab` `wget` `ulimit`
# Linux Shell commands
Chang's collection of working examples in Bash, Shell
---
## awk
Syntax `awk -F "$file_delimiter" -v awk_var1=${shell_var1} -v awk_var2=${shell_var2} 'BEGIN {print "colname1","colname2"} {print $awk_var1,$awk_var2}' $filePath`
### Loop thruough unique combinations (e.g. lower diagonal) of a 10 by 10 matrix using `for` and `awk`.
```r
# You should know how many iterations will be run, say 10choose2. Get this in R
choose(10,2)
[1] 45
```
```bash=
# Location of a tsv file to loop thru
filePath_munged_GWAS=${loc_LDSC_input}/file-info_munged-QCed-GWASs.tsv
# Get number of lines including header row
num_lines=$(awk -F"\t" 'END {print NR}' $filePath_munged_GWAS) # 11
#-------------------------------------------------------------------
# Loop through unique combinations of 10 munged files
## Number of iterations: 45 (10 choose 2)
#-------------------------------------------------------------------
# Loop thru 45 combinations
## Skip first line (header)
count=0
for ((i=2;i <=${num_lines}-1;i++));do
for ((j=$i+1;j<=${num_lines};j++));do
count=$(($count+1));
jobName="rG_iteration_$count"
echo "==================================== Iteration $count ======================================================="
#---------------------------------------------------------------------------------------------------
# Assign values of field 1-3 per two line counter variables $i and $j of the input file to variables
#---------------------------------------------------------------------------------------------------
file=${filePath_munged_GWAS}
mungedFile_1_field_1=$(awk '(NR=='$i') {print $1}' $file); # file path
mungedFile_1_field_2=$(awk '(NR=='$i') {print $2}' $file); # consortium
mungedFile_1_field_3=$(awk '(NR=='$i') {print $3}' $file); # trait
mungedFile_1_field_9=$(awk '(NR=='$i') {print $9}' $file); # population prevalence for binary traits
mungedFile_1_field_10=$(awk '(NR=='$i') {print $10}' $file); # sample prevalence for binary traits
mungedFile_2_field_1=$(awk '(NR=='$j') {print $1}' $file); # file path
mungedFile_2_field_2=$(awk '(NR=='$j') {print $2}' $file); # consortium
mungedFile_2_field_3=$(awk '(NR=='$j') {print $3}' $file); # trait
mungedFile_2_field_9=$(awk '(NR=='$j') {print $9}' $file); # population prevalence for binary traits
mungedFile_2_field_10=$(awk '(NR=='$j') {print $10}' $file); # sample prevalence for binary traits
echo "munged file 1: ${mungedFile_1_field_2}-${mungedFile_1_field_3}, ${mungedFile_1_field_1}, ${mungedFile_1_field_9}, ${mungedFile_1_field_10}"
echo "munged file 2: ${mungedFile_2_field_2}-${mungedFile_2_field_3}, ${mungedFile_2_field_1}, ${mungedFile_2_field_9}, ${mungedFile_2_field_10}"
#-------------------------------------------------------------------------------------------------------------
# Calculate genetic correlation between munged file 1 and munged file 2 where the 2 files are different traits
#-------------------------------------------------------------------------------------------------------------
qsub -N $jobName -v v_MF_1_filePath=${mungedFile_1_field_1},v_MF_1_consortium=${mungedFile_1_field_2},v_MF_1_trait=${mungedFile_1_field_3},v_MF_1_population_prevalence=${mungedFile_1_field_9},v_MF_1_sample_prevalence=${mungedFile_1_field_10},v_MF_2_filePath=${mungedFile_2_field_1},v_MF_2_consortium=${mungedFile_2_field_2},v_MF_2_trait=${mungedFile_2_field_3},v_MF_2_population_prevalence=${mungedFile_2_field_9},v_MF_2_sample_prevalence=${mungedFile_2_field_10},v_folderPath_output=${loc_LDSC_rG} -l ncpus=${num_cpu},walltime=${runTime_requested},mem=${memory_requested} -e ${pbs_output_dir}/${jobName}.pbs.err -o ${pbs_output_dir}/${jobName}.pbs.out ${filePath_jobScript};
done
done
```
---
### Count number of lines of a file with header
[Get the line count from 2nd line of the file](https://stackoverflow.com/questions/40689719/get-the-line-count-from-2nd-line-of-the-file)
```bash=
# Use awk END block to count number of records
awk -F"\t" 'END {print NR-1}' $filePath_GWAS_info
# Use tail and wc -l
tail -n +2 file | wc -l
```
---
### Matching patterns in `awk`
[AWK: How to Compare Two Variables with Regular Expression](https://unix.stackexchange.com/questions/421404/awk-how-to-compare-two-variables-with-regular-expression)
```bash=
# Example 1: matching a pattern defined by a built-in variable
## A file to work on
filePath=/mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/LD-score-correlation/input/header-fieldNumber-colname_ICC-CI
cat ${filePath}
1 SNP
2 Allele1
3 Allele2
4 MAF
5 Effect
6 StdErr
7 P
8 N
## The pattern to search/match in the file.
colname_p_value="P"
## Find value of column 1 corresponding to the pattern using full word matching. Note "P" is also found in "SNP". Without full word matching, this returned 1 and 7.
awk -v pattern=${colname_p_value} '{if ($2 ~ "^" pattern "$") print $1}' ${filePath}
7
# Example 2: matching a pattern using regular expression. Here you cannot use a built-in vairable inside the regex. The regex should be just a string (i.e. hardcoding text)
awk '{if ($2 ~ /^P$/) print $1}' ${filePath}
7
```
---
### Display standard output or a file delimited by one or more whitespaces as one column using `awk -F" +"`. It's dangerous to list files from multiple folders using a single `ls -l`, as files will be sorted rather than the order you put after `ls -l` . If you need just file paths, use `realpath`
* [awk -F“ ” delimit by space greater than 1](https://unix.stackexchange.com/questions/399197/awk-f-delimit-by-space-greater-than-1)
```bash=
# The standard output to manipulate to show just the last column (file name here)
## The delimiters are one or more spaces
ls -l *_noICC_LDWindow-kb-10000_R2-0.01_p1-*_p2-*.clumped | head -2
-rw-r--r-- 1 lunC sec_NickM 10577 Apr 3 10:08 ai_noICC_LDWindow-kb-10000_R2-0.01_p1-1e-5_p2-1e-5.clumped
-rw-r--r-- 1 lunC sec_NickM 201 Apr 3 10:08 ai_noICC_LDWindow-kb-10000_R2-0.01_p1-5e-8_p2-1e-6.clumped
# Read the stdout using awk
ls -l *_noICC_LDWindow-kb-10000_R2-0.01_p1-*_p2-*.clumped | awk -F" +" '{print $9}' | head -2
ai_noICC_LDWindow-kb-10000_R2-0.01_p1-1e-5_p2-1e-5.clumped
ai_noICC_LDWindow-kb-10000_R2-0.01_p1-5e-8_p2-1e-6.clumped
```
---
### Duplicate or repeat each line for a known number of times
*[Repeat each line multiple times](https://unix.stackexchange.com/questions/81904/repeat-each-line-multiple-times)
```bash=
## A file with 3 lines
cat file
a
b
c
## Repeat each line for 4 times
awk '{for(i=0;i<4;i++)print}' file
```
---
### Count frequency of unique values in a column
```bash=
# Count (field 1) and unique value (field 2) of column 11 (NF=11, $NF means $11)
awk '{print $NF}' Cannabis_ICC_UKB.txt | sort | uniq -c
771056 149468
111811 162082
17267 184765
4128890 22683
611134 35297
5895434 57980
1 N
```
---
### Count number of lines with different column numbers
* [Getting the count of unique values in a column in bash](https://stackoverflow.com/questions/4921879/getting-the-count-of-unique-values-in-a-column-in-bash)
```bash=
# Output number of lines (field 1) and column numbers (field 2)
awk '{print NF}' Cannabis_ICC_UKB.txt | sort| uniq -c
900134 10
10635459 11
```
---
### Pass shell variables to awk variables. An example with `awk -v` `awk BEGIN{}` and `awk {action}`
* [How do I use shell variables in an awk script?](https://stackoverflow.com/questions/19075671/how-do-i-use-shell-variables-in-an-awk-script)
```bash=
# The example on the website
awk -v a="$var1" -v b="$var2" 'BEGIN {print a,b}'
```
---
### Use awk -v variables in the search pattern. Unlike shell variables, awk variables are not prefixed with `$`. You can't embed them inside a `/regex constant/` it's just text in there.
* [How to pass shell variable to an awk search pattern? \[duplicate\]](https://unix.stackexchange.com/questions/216464/how-to-pass-shell-variable-to-an-awk-search-pattern)
```bash=
# part 1 : searching with 3 patterns without awk variables
awk '{if ($1 ~/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1/ && $1 ~/GWAS-UKB/ && $1 ~ /dosageFam_Release8_HRCr1.1/) print $0}' ${locASCOut}/filePath_summedRiskProfiles.S1-S8_newHeader > ${locASCTest}/filePath_group1
# part 2 : searching with 3 patterns with awk variables
$ p1=innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1
$ p2=GWAS-UKB
$ p3=dosageFam_Release8_HRCr1.1
$ awk -v v_p1=$p1 -v v_p2=$p2 -v v_p3=$p3 '{if ($1 ~ v_p1 && $1 ~ v_p2 && $1 ~ v_p3) print $0}' ${locASCTest}/filePath_group1
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ASS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ES/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NCD/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NSDPW/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-SS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
```
---
### Using shell variables in awk print
```bash=
# cut doesn't support ordering of the fields so the following code won't give you the order
cut -d" " -f1,2,${fNum},8,10-12,85-88 $phenoRaw | grep -v FAMID > $outputFilePath;
# use awk print with a awk variable that is assigned from a shell variable
fNum=21;
awk -v fieldPosPRS=${fNum} 'BEGIN{OFS=IFS=" "} {print $1,$2,$fieldPosPRS,$8,$10,$11,$12,$85,$86,$87,$88}' $phenoRaw | grep -v FAMID > $outputFilePath;
```
---
### Extract field names from field number with a 2 column file (field1=filed number; field2=filed name)
```bash=
# Save field position and field names as a 2 column file
## grep words that start with UKB, leaving first line out
## \b : beginning of a word
sh $locScripts/fileChecker.sh $phenoRaw | grep -E '\bUKB' > qcovar_fNum_fNam
# Use field 1 of the file as iterators
# Use awk to extract field 2 per value of field 1
for i in `cat qcovar_fNum_fNam | cut -d" " -f1`; do
fNum=${i};
fNam=`awk -v fieldPos=${i} '($1==fieldPos){print $2}' qcovar_fNum_fNam`;
outputFileName=qcovar_PRS_${fNam};
outputFilePath=${locQCovar_r_everDrugs}/${outputFileName};
echo "fNum=$fNum";
echo "fNam=$fNam";
echo "outputFileName=$outputFileName";
echo "outputFilePath=$outputFilePath";
# Column-subset FAMID, ID and a qcovar field
cut -d" " -f1,2,${fNum} $phenoRaw | grep -v FAMID > $outputFilePath;
done
```
---
### Compare two files by merging them and checking the ratio of two numeric fields =1 or not
```bash=
paste -d " " $mainFolder/F02_PRScalc/SCZ.S1.rawSum /mnt/lustre/working/lab_nickm/lunC/PRS_test/test/summedRiskProfiles.SCZ2.S1 |awk '(($1<-0.00001)||($1>0.00001)){print $1,$5,$5/$1}'|sort -g -k3|tail
2.03784 2.03784 1
2.04477 2.04477 1
2.08924 2.08924 1
2.10368 2.10368 1
2.11138 2.11138 1
2.16875 2.16875 1
2.1767 2.1767 1
2.22143 2.22143 1
2.26245 2.26245 1
-5.39526e-05 -5.39526e-05 1
```
---
### Search lines ended with colon with awk, replace ending colon with nothing, then replace newline with a space
* [list lines ending with a period or semicolon](https://stackoverflow.com/questions/12707921/list-lines-ending-with-a-period-or-semicolon)
* [Replace comma with newline in sed](https://stackoverflow.com/questions/10748453/replace-comma-with-newline-in-sed)
* [How to remove a character at the end of each line in unix](https://stackoverflow.com/questions/14840953/how-to-remove-a-character-at-the-end-of-each-line-in-unix)
```bash=
# A file from listing two subfolders
cat dirList_uniqSNPs_allUKBs_Release8
uniqSNPs_allUKBs_Release8-1000GPhase3:
innerJoinedSNPsByCHRBP_metaDataQCed_Release8_1000GPhase3_AND_GWASQCedChrConcat_UKB1249_pastTobaccoSmoking
innerJoinedSNPsByCHRBP_metaDataQCed_Release8_1000GPhase3_AND_GWASQCedChrConcat_UKB1498_coffeeIntake
uniqSNPs_allUKBs_Release8-HRCr1.1:
innerJoinedSNPsByCHRBP_metaDataQCed_Release8_1000GPhase3_AND_GWASQCedChrConcat_UKB1249_pastTobaccoSmoking
innerJoinedSNPsByCHRBP_metaDataQCed_Release8_1000GPhase3_AND_GWASQCedChrConcat_UKB1498_coffeeIntake
# Search lines ended with colon with awk
awk '/[:]$/' dirList_uniqSNPs_allUKBs_Release8
# Search lines ended with colon with grep
grep '[:]$' dirList_uniqSNPs_allUKBs_Release8
# Search lines ended with colon with awk, replace ending colon with nothing, then replace newline with a space
awk '/[:]$/' dirList_uniqSNPs_allUKBs_Release8 | sed 's/:$//' | tr "\n" " "
uniqSNPs_allUKBs_Release8-1000GPhase3 uniqSNPs_allUKBs_Release8-HRCr1.1
```
---
### Write a header line, multiple if conditions with awk
```bash=
# Filter out SNPs with multiple criteria
## FS: input file field separator. OFS: output file field separator.
## Print "colA","colB".... : add a line of text before file is read
## if statement must be contained within the curly braces
## ~ : matches a regex. !~ not match a regex
## /[0-9]*:[0-9]*:[0-9]*/ : pattern as anyNumberAnyLength:anyNumberAnyLength:anyNumberAnyLength (CHR:BP:version)
## length($2)== 1 && length($3)== 1 : removed reference alleles and alternative alleles with > 1 allele
awk ' BEGIN { FS="\t"; OFS=" "; print "CHR:BP","REF","ALT","bp_Build37","SNP_dbSNP", "MAF","Rsq_rederived" } {if($1 !~ /[0-9]*:[0-9]*:[0-9]*/ && length($2)== 1 && length($3)== 1 && $18 ~/rs/ && $44>=0.01 && $44<=0.99 && $48>=0.6) print $1,$2,$3,$17,$18,$44,$48} ' $meta1000G
```
---
### Two-file processing in `awk first_file second_file` In awk, `FNR` refers to the **record number (typically the line number) in the current file** and `NR` refers to the **total record number**. The operator == is a comparison operator, which returns true when the two surrounding operands are equal. This means that the condition `NR==FNR` is only true for the first_file, as FNR resets back to 1 for the first line of each file but NR keeps on increasing. This pattern is typically used to perform actions on only the first file. The next inside the block means any further commands are skipped, so they are only run on files other than the first. The condition `FNR==NR` compares the same two operands as `NR==FNR`, so it behaves in the same way.
* [What is “NR==FNR” in awk?](https://stackoverflow.com/questions/32481877/what-is-nr-fnr-in-awk)
```bash=
# (NR!=1) prevents reading first row (header) of file 2
# If field 3 of file2 ${gwas_file1} matches field1 of file1 ${ref_snplist}, then output field 3,5,4,8,9,7
# If field 3 of file2 ${gwas_file1} doesn't matches field1 of file1 ${ref_snplist}, then skip the following actions
awk 'BEGIN {print "SNP EA NEA BETA SE P"}(NR==FNR)&&(NR!=1){a[$1];next;}{if($3 in a) print $3,$5,$4,$8,$9,$7}' ${ref_snplist} ${gwas_file1} > ${ldsc_gwas1}
```
---
### Search for a pattern, filtering out a column that ends with number
```bash=
# $2 !~ /[0-9]$/ : filter out field2 that ends with a number 0- 9
## /search-pattern/ [0-9] means number
## $ means end with, ^ means start with
wc -l GWASQCedChrConcat_UKB* | awk '$2 !~ /[0-9]$/'
25822798 GWASQCedChrConcat_UKB1249_pastTobaccoSmoking
25822798 GWASQCedChrConcat_UKB1498_coffeeIntake
77509877 GWASQCedChrConcat_UKB1558_alcoholIntakeFreq
25822798 GWASQCedChrConcat_UKB20453_everTakenCannabis
25822798 GWASQCedChrConcat_UKB20455_ageLastTookCannabis
77509877 GWASQCedChrConcat_UKB21001_BMI
25822798 GWASQCedChrConcat_UKB3456_numberCigarettesDaily
77509877 GWASQCedChrConcat_UKB50_standingHeight
361643819 total
```
---
### Count number of fields, total number of lines with awk and tail
```bash=
# a file to analyse
file=UKB50_lineNum;
cat $file;
# Count NF, total record number with awk and tail
## awk -v fileName=$file: create a variable in awk named fileName. This takes value from the shell variable dollar-signfile
## BEGIN{}: A BEGIN rule is executed once only, before the first input record is read
## OFS: awk built-in variable for output field separator
## END{}: an END rule is executed once only, after all the input is read. This is usually used when adding a summmary line to the end of the file or performing calculation.
## NR: awk built-in variable for record number
## NF: awk built-in variable for number of fields
## `tail -n 1` prints only the last line. Otherwise, every line of the file will be printed
awk -F" " -v fileName=$file 'BEGIN{OFS=",";} END{print NR OFS NF OFS fileName}' $file | tail -n 1
22 2 UKB50_lineNum
'BEGIN{OFS=" ";} {print $3,$4;}'
```
---
### print duplicates with awk
* [Print the duplicate lines in a file using awk](https://stackoverflow.com/questions/36506212/print-the-duplicate-lines-in-a-file-using-awk)
```bash=
# A test data file with duplicates $1
cat tmp1
a b c d e
1 2 3 4 5
1 6 7 8 9
2 3 4 5 6
# Print only the lines with duplicated $1
awk 'seen[$1]++' tmp1
1 6 7 8 9
# Print lines with duplicated field 1 values and their previous lines
# An example of two file processing where 1st file tmp1 is used as a filter for 2nd file tmp1
# If you want to stick with just plain awk, you'll have to process the file twice
## once to generate the counts,
## once to eliminate the lines with count equal 1 (lines with unique $1):
awk 'NR==FNR {count[$1]++; next} count[$1]>1' tmp1 tmp1
1 2 3 4 5
1 6 7 8 9
```
---
### Remove all occurences of duplicated lines
```bash=
# dupicated $1 in line2
[lunC@hpcpbs01 test]$ cat tmp1
a b c d e
1 2 3 4 5
1 6 7 8 9
2 3 4 5 6
# Remove line1 and line2
[lunC@hpcpbs01 test]$ awk '{seen[$1]++; a[++count]=$0; key[count]=$1} END {for (i=1;i<=count;i++) if (seen[key[i]] == 1) print a[i]}' tmp1
a b c d e
2 3 4 5 6
```
---
### Count columns and rows of a file
* [count number of columns in bash](https://stackoverflow.com/questions/5761212/count-number-of-columns-in-bash)
```bash=
inputFile=mergedSummedPRSOrderedIDremapped.S1-S8.ADHD2017_ASD2015_SCZ2
# count number of columns using awk
awk '{print NF}' $inputFile | sort -nu | tail -n 1
# Count rows
cat $inputFile | wc -l
27462
```
---
### `awk -v` The -v option allows shell variables to pass into your awk script.
* [awk -v](http://www.unix.com/unix-for-dummies-questions-and-answers/15651-awk-v.html)
```bash=
Bob="Hello"
Sue="There"
awk -vFred=$Bob -vJoe=$Sue ........rest of awk statement ...........
```
---
### List position of every field name of a .gz file with grep and awk
```bash=
# Suppose header row contains the string CHR
zcat /working/lab_nickm/lunC/GWAS_summary/adhd_jul2017.gz | grep CHR | awk '{for (i=1; i <=NF; i++) print i, $i}'
1 CHR
2 SNP
3 BP
4 A1
5 A2
6 INFO
7 OR
8 SE
9 P
```
---
## Parameter expansion
### Remove the last or all the file extensions `( .*)` using parameter expansion [How can I remove the extension of a filename in a shell script?](https://stackoverflow.com/questions/12152626/how-can-i-remove-the-extension-of-a-filename-in-a-shell-script)
```bash
filename=foo.txt.csv
#Remove the last file extension using parameter expansion
echo "${filename%.*}"
foo.txt
#Remove all the file extensions using parameter expansion
echo "${filename%%.*}"
foo
```
---
## Variable expansion
* [Expansion of variable inside single quotes in a command in Bash](https://stackoverflow.com/questions/13799789/expansion-of-variable-inside-single-quotes-in-a-command-in-bash)
```bash!
line_read_from=63
line_read_until=64
# Correct usage. The 2 variables are expaned within double quotes
sed -n "${line_read_from},${line_read_until}p" filePath
# Wrong usage. The 2 variables are NOT expanded within single quotes
sed -n '${line_read_from},${line_read_until}p' filePath
# sed: -e expression #1, char 4: extra characters after command
# Correct usage.
sed -n '63,64p' filePath
sed -n "63,64p" filePath
```
---
## echo
### How to echo an expression that contains single quotes, dobule quotes, and variables? If you will `echo "expression"`, escape double quotes within and dollar signs for variables that need not to expand (e.g. awk variables). Don't escape dollar signs for variables that need to expand (e.g. output folder paths) [How do I echo an expression with both single and double quotes?](https://unix.stackexchange.com/questions/133958/how-do-i-echo-an-expression-with-both-single-and-double-quotes)
```bash!
# An expression to echo is
awk -F"\t" -v fieldPosSNPID=$field_RSNum 'NR==FNR {count[$fieldPosSNPID]++;next} count[$fieldPosSNPID]>1' ${filePath} ${filePath} > ${output_folder_path}/${fileName}.allOccurrenceDup
# First, comment out the expression so it won't get executed during code testing
# Next, use double quotes to quote the expression.
# Finally, escape every double quote, and awk variables in the expression
filePath="/home/script"
output_folder_path="/working/output"
fileName="test"
echo "awk -F\"\t\" -v fieldPosSNPID=\$field_RSNum 'NR==FNR {count[\$fieldPosSNPID]++;next} count[\$fieldPosSNPID]>1' ${filePath} ${filePath} > ${output_folder_path}/${fileName}.allOccurrenceDup"
# awk -F"\t" -v fieldPosSNPID=$field_RSNum 'NR==FNR {count[$fieldPosSNPID]++;next} count[$fieldPosSNPID]>1' /home/script /home/script > /working/output/test.allOccurrenceDup
```
---
### How to echo an expression with either single or double quotes, not mixture?
```bash!
# Wrong: double quotes wihtin double quotes
echo " cut -d" " -f1 file"
# cut -d -f1 file
# Correct: double quotes are escaped wihtin double quotes
echo " cut -d\" \" -f1 file"
# Wrong: single quotes wihtin single quotes
echo ' cut -d' ' -f1 file'
# cut -d -f1 file
# Wrong: single quotes are escaped wihtin single quotes
echo ' cut -d\' \' -f1 file'
# >
# Correct: single quotes wihtin double quotes
echo " cut -d' ' -f1 file"
# cut -d' ' -f1 file
# Correct: double quotes within single quotes
echo ' cut -d" " -f1 file'
#cut -d" " -f1 file
```
---
### When you read a CSV file and `echo` doesn't print variable values correctly, it's likely due to Windows special character not readable to Linux. `dos2unix` the CSV file before reading it
---
### Be aware `echo $var` changes separators to white space. Use `echo "$var"` to keep the original separator unchanged
[Echo changes my tabs to spaces](https://stackoverflow.com/questions/5127954/echo-changes-my-tabs-to-spaces)
```bash!
## Change P_BOLT_LMM to pvalue
filePath_UKB_CPPD="${locUKB_CCPD_QC3}/QCed-GWAS-UKB-CCPD_headed";
header_new=$(head -1 ${filePath_UKB_CPPD} | sed 's/P_BOLT_LMM/pValue/g')
## Correct use of echo
cat <(echo "$header_new") <(tail -n +2 $filePath_UKB_CPPD)
## Wrong use of echo. $header_new separators changed to white space whereas the tail part is tab-separated
cat <(echo $header_new) <(tail -n +2 $filePath_UKB_CPPD)
```
---
### Use echo to print commands to execute per iteration. Copy the commands on the stdout and run them. This allows you to run just one iteration of code for testing. The `echo "$locLDSC/munge_sumstats.py..."` is exactly the same as the command in the next line
```bash!
# Loop thru each line of the comma-separated GWAS information file, skipping 1st row (header)
## Number of iteration: 8
count=0;
for line in `tail -n +2 $filePath_csv`;do
count=$((${count}+1));
echo "========================iteration $count =========================";
filePath=`echo $line | cut -d"," -f1`; # get column 1
fileName=`basename ${filePath}`;
sampleSize=`echo $line | cut -d"," -f2`; # get column 2
allele1=`echo $line | cut -d"," -f3`; # get column 3
allele2=`echo $line | cut -d"," -f4`; # get column 4
pValue=`echo $line | cut -d"," -f5`; # get column 5
echo "filePath=${filePath}";
echo "fileName=${fileName}";
echo "sampleSize=${sampleSize}";
echo "allele1=${allele1}";
echo "allele2=${allele2}";
echo "pValue=${pValue}";
echo "$locLDSC/munge_sumstats.py --sumstats ${filePath} --N ${sampleSize} --out ${loc_rG_munged}/${fileName}.munged --merge-alleles ${filePath_SNPList} --a1 ${allele1} --a2 ${allele2} --p ${pValue}";
# Munge GSCAN GWAS files
#$locLDSC/munge_sumstats.py --sumstats ${filePath} --N ${sampleSize} --out ${loc_rG_munged}/${fileName}.munged --merge-alleles ${filePath_SNPList} --a1 ${allele1} --a2 ${allele2} --p ${pValue} ;
done
```
---
### Use echo to bebug code. Add echo "some text" after the line that may not work. If the echo prints something in the output script.sh.o* then the line is working.
```bash!
# If you are not sure if the if statement is working, add echo after it
if [ $dataSource = UKB ]; then
echo "this file is from UKB"
#awk -F"\t" 'NR==FNR {count[$1]++;next} count[$1]>1' $filePath $filePath > $folderPath/QC1_find_allOccurencesOfDuplicatedSNPs/${fileName}_allOccurrenceDup
else
echo "this file is NOT from UKB"
fi
# Check if echo correctly print something
less PRS_UKB_201711_step01-04_QC_combine_GWAS_jobScript.sh.o4833530
```
---
## cat
### Use new line character in a heredoc
[\n in variable in heredoc](https://stackoverflow.com/questions/28090477/n-in-variable-in-heredoc)
```bash=
# This won't work
(cat <<- _EOF_
word1\nword2\nword3
_EOF_
) > my-heredoc
# This works
newline=$'\n'
(cat <<- _EOF_
word1${newline}word2${newline}word3
_EOF_
) > my-heredoc
```
---
### Use variable inside a here document. Variable expansion is the default behavior inside of here-docs. You disable that behavior by quoting the label (with single or double quotes).
* [Using variables inside a bash heredoc](https://stackoverflow.com/questions/4937792/using-variables-inside-a-bash-heredoc)
```bash=
# Write file paths of QCed GWAS files and their subset files into another file for iterations
(cat <<- _EOF_
GWAS_file_path SNP_PValue_file_path
$locUKB3456_QC3/QCed-GWAS-UKB3456_headed $locUKB3456_input/SNP_PValue_QCed-GWAS-UKB3456_headed
$locUKB_ESDPW_QC3/QCed-GWAS-UKB-ESDPW_headed $locUKB_ESDPW_input/SNP_PValue_QCed-GWAS-UKB-ESDPW_headed
$locUKB_CCPD_QC3/QCed-GWAS-UKB-CCPD_headed $locUKB_CCPD_input/SNP_PValue_QCed-GWAS-UKB-CCPD_headed
$locUKB20161_QC3/QCed-GWAS-UKB-PYOS_headed $locUKB20161_input/SNP_PValue_QCed-GWAS-UKB-PYOS_headed
_EOF_
) > $locMR/filePath_QCed-UKB-GWAS_SNP-PValue-subset.txt
# Check your here document
head -2 $locMR/filePath_QCed-UKB-GWAS_SNP-PValue-subset.txt
GWAS_file_path SNP_PValue_file_path
/mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/data/UKB3456-numCigareDaily_IID-NA-in-UKB204534-everUsedCannabis/QC3_remove_ambiguousSNPs_indel/QCed-GWAS-UKB3456_headed /mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/data/UKB3456-numCigareDaily_IID-NA-in-UKB204534-everUsedCannabis/LD-based_SNP_clumping/input/SNP_PValue_QCed-GWAS-UKB3456_headed
```
---
### Create a here document with some space-delimited data
* [How can I write a heredoc to a file in Bash script?](https://stackoverflow.com/questions/2953081/how-can-i-write-a-heredoc-to-a-file-in-bash-script)
```bash=
locInput=/mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/data/noICC_results/LD-based_SNP_clumping/input;
mkdir -p $locInput;
# Create a here document with 3 rows
## row1: header
## row2 to last row: each clumping criteria
cd $locInput;
(cat <<- _EOF_
r2 LDWindow p1 p2
0.01 10000 5e-8 1e-6
0.01 10000 1e-5 1e-5
_EOF_
) > ${locInput}/LD-SNP-clumping-criteria.txt
```
---
### Use - as the stdin for cat
* [Usage of dash (-) in place of a filename](https://unix.stackexchange.com/questions/16357/usage-of-dash-in-place-of-a-filename)
```bash
# - on the right of cat is used as stdin (the stdout from paste)
paste -d"," ${locASCTest}/temp ${locASCTest}/fileJoinerOptions_non-directory-part2 | cat - ${locASCTest}/fileJoinerOptions_non-directory-part3 > ${locASCTest}/${p1Dash}_${p2}_${p3Dash};
```
---
## touch
### Create a new, empty file using `touch` command
* [The touch Command](http://www.linfo.org/touch.html)
```bash=
touch /mnt/backedup/home/lunC/scripts/MR_ICC_GSCAN_201806/MR_step04-01-02_LD-clumping-criteria.sh
```
---
## cut
### split a file by byte position, character or delimiter
`cut -d` is used to specify the delimiter (default=tab)
`cut -f` specify the field that should be cut, allowing use of a field range
[cut](https://shapeshed.com/unix-cut/#what-is-the-cut-command-in-unix)
---
### Subset 2nd column of a tab-delimited file using `cut`.
*[How to define 'tab' delimiter with 'cut' in BASH?](https://unix.stackexchange.com/questions/35369/how-to-define-tab-delimiter-with-cut-in-bash)
```bash=
# -d'\t' returned an error cut: the delimiter must be a single character
# use -d$'\t' instead
cut -d$'\t' -f2 $dir_main/$gwasInfo
```
---
## paste
### displays the corresponding lines of multiple files side-by-side
[paste](https://www.computerhope.com/unix/upaste.htm)
`paste -d` is used to specify the delimiter in the output file (default=tab)
---
### `cut` doesn't support ordering of fields. Reorder columns of a space-delimited file using cut and paste or awk
old column order: `$1,$2......$19`
new column order: `$2,$1,$3...$19`
```bash
# divide the file into 2 temp files
awk '{print $2,$1}' PRS.S1-S8.ADHD2017_ASD2015 > temp_f2-1 && cut -d ' ' -f3-19 PRS.S1-S8.ADHD2017_ASD2015 > temp_f3-19
# Horizontally combine the 2 temp files. Use space as the delimiter in the output file
paste -d' ' temp_f2-1 temp_f3-19 > t_PRS.S1-S8.ADHD2017_ASD2015
```
```bash=
# Another example
cut -d" " -f2 summedPRSS1-S8UKBAllPheno_PCs_impCov > temp1;
cut -d" " -f1,3-78 summedPRSS1-S8UKBAllPheno_PCs_impCov > temp2;
paste -d" " temp1 temp2 > temp3;
# Do this as a oneliner
paste -d" " <(cut -d" " -f2 summedPRSS1-S8UKBAllPheno_PCs_impCov) <(cut -d" " -f1,3-78 summedPRSS1-S8UKBAllPheno_PCs_impCov) > temp1
```
---
## Special characters
### Escape `$var` within single quotes that are enclosed by double quotes by `\` if you don't want it to be interpreted. The ' single quote character in your echo example gets it literal value (and loses its meaning) as it enclosed in double quotes ("). The enclosing characters are the double quotes.
[Single quote within double quotes and the Bash reference manual](https://unix.stackexchange.com/questions/169508/single-quote-within-double-quotes-and-the-bash-reference-manual)
```bash=
# Commands to test by echo
awk '{print $3}' $filePath_left_table | sed '/^ *$/d' > ${output_folder}/clumped-SNPs_${fileName_fileRight_part1} ;
# Print the commands using echo
## Not to be interpreted: $3, sed `/^*$/d`
## To be interpreted: $filePath_left_table, ${output_folder},${fileName_fileRight_part1}
echo "awk '{print \$3}' $filePath_left_table | sed '/^ *\$/d' > ${output_folder}/clumped-SNPs_${fileName_fileRight_part1}"
awk '{print $3}' /mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/data/UKB-estimated-standard-drinks-per-week_IID-NA-in-UKB204534-everUsedCannabis/LD-based_SNP_clumping/output/GWAS-UKB-ESDPW_LDWindow-kb-10000_R2-0.01_p1-5e-8_p2-1e-6.clumped | sed '/^ *$/d' > /mnt/lustre/working/lab_nickm/lunC/MR_ICC_GSCAN_201806/data/ICC-cannabis-ever/QC4_GWAS_from_clumped_SNPs/clumped-SNPs_QCed-GWAS-UKB-ESDPW_headed
```
---
## sed
### **sed** performs string-based manipulation on an input stream. **tr** command Translate, squeeze, delete characters from standard input, writing to standard output
---
### Delete/Remove punctuation marks from a string
[Removing all special characters from a string in Bash](https://stackoverflow.com/questions/36926999/removing-all-special-characters-from-a-string-in-bash/36930095)
```bash!
# Get the line to parse
sed -n "${line_h2_pheno1}p" < $filePath
#Total Liability scale h2: 0.0325 (0.0044)
# Extract heritability estimate and standard error of trait 1
## tr -d deletes the character sets defined in the arguments on the input stream
## [:punct:] includes characters ! " # $ % & ' ( ) * + , - . / : ; < = > ? @ [ \ ] ^ _ ` { | } ~
## This deletes decimal dot, which is not what I want
sed -n "${line_h2_pheno1}p" $filePath | sed -e 's/Total Liability scale h2: //g' | tr -d "[:punct:]"
00325 00044
# Extract heritability estimate and standard error of trait 1 and replace '(' and ')' with nothing
sed -n "${line_h2_pheno1}p" $filePath | sed -e 's/Total Liability scale h2: //g;s/(//g;s/)//g'
0.0325 0.0044
```
---
### Remove leading blanks or tabs in every line
[sed tip: Remove / Delete All Leading Blank Spaces / Tabs ( whitespace ) From Each Line](https://www.cyberciti.biz/tips/delete-leading-spaces-from-front-of-each-word.html)
```bash
# To remove all whitespace (including tabs) from left to first word, enter:
sed -e 's/^[ \t]*//' file
## -e : expect sed command to use regular expression
## s/ : Substitute command ~ replacement for pattern (^[ \t]*) on each addressed line
## ^[ \t]* : Search pattern ( ^ – start of the line; [ \t]* match one or more blank spaces including tab)
## // : Replace (delete) all matched pattern
```
---
### Replace . with ImpCov in first line
```bash
# Replace . with ImpCov with sed '1 s/\./ImpCov/'
## 1 part only replaces the first line
## needs to escape special character . from being interpreted
sed '1 s/\./ImpCov/' temp1_IDremapped > summedPRSS1-S8UKBAllPheno_PCs_impCov_IDRemapped
```
---
### Insert text before the first line of a file using sed
syntax `sed -i '1i text' filename`
* `-i` option stands for "in-place" editing. It is used to modify the file without the need to save the output of sed command to some temporary file and then replacing the original file.
* `1i` or `1 i` 1 is to select first line. i means inserting text and newline
```bash=
# Add header row to the meta data
sed -i '1iSNP REF ALT bp_Build37 SNP_dbSNP MAF Rsq_rederived' metaDataQCed_Release8_1000GPhase3;
sed -i '1iSNP REF ALT bp_Build37 SNP_dbSNP MAF Rsq_rederived' metaDataQCed_Release8_HRCr1.1;
```
---
### Get Nth line
`sed -n '2p' < file.txt` will print 2nd line
`sed -n '2011p' < file.txt` will print 2011th line
`sed -n '10,33p' < file.txt` prints line 10 up to line 33
`sed -n '1p;3p' < file.txt` prints 1st and 3th line
`sed 'NUMq;d' file` Where NUM is the number of the line you want to print; so, for example, sed '10q;d' file to print the 10th line of file.
Explanation:
NUMq will quit immediately when the line number is NUM.
d will delete the line instead of printing it; this is inhibited on the last line because the q causes the rest of the script to be skipped when quitting.
If you have NUM in a variable, you will want to use double quotes instead of single: `sed "${NUM}q;d" file`
[Bash tool to get nth line from a file](https://stackoverflow.com/questions/6022384/bash-tool-to-get-nth-line-from-a-file)
```bash=
# get line 21759 up to 21762
sed -n '21759,21762p' < /mnt/lustre/working/lab_nickm/lunC/PRS_test/test/clumpedSNPs.SCZ2.1
rs17025931
```
---
### Substitutes "dump" for "C G", "G C", "T A", and "A T" These are ambiguous SNPs
```bash
sed 's/C G/dump/g;s/G C/dump/g;s/T A/dump/g;s/A T/dump/g' temp4 > temp5
```
---
## tr
### sed versus tr
[Difference Between tr and sed Command](https://www.linux.com/forums/command-line/difference-between-tr-and-sed-command)
---
### replace `+` with white-space ' ' and this type of replacement can be done with both tr as well sed command as below
```bash=
echo This+is+test+for+tr+and+sed |tr '+' ' '
#This is test for tr and sed
echo This+is+test+for+tr+and+sed |sed 's/\+/ /g'
#This is test for tr and sed
```
---
### Deleting 'chr' with sed. As this edits multiple characters, use `sed` instead of `tr`
```bash=
string1="chr1"
# undesired use of tr
echo $string1 | tr 'chr' ' '
1
# desired use of sed
echo $string1 | sed 's/chr/ /g'
1
```
---
### `tr` has done character based transformation and it is replacing good to best as g=b, o=e, o=s, d=t and because o is double so it ignores the first rule and using o=s.
```bash!
echo I am a good boy | tr 'good' 'test'
#I am a tsst bsy
```
---
### But `sed` is string based transformation and if there will 'good' string more than one time those will replace with 'best'
```bash!
echo I am a good boy | sed 's/good/best/g'
#I am a best boy
```
---
## for
### `for` loop is a block of code that iterates through a list of commands as long as the loop control condition is true. During each pass through the loop, arg takes on the value of each successive variable in the list
```bash!
for arg in [list]
do
command(s)...
done
```
---
### Loop through a range of numbers
[How to iterate Bash for Loop variable range under Unix or Linux](https://www.cyberciti.biz/faq/unix-linux-iterate-over-a-variable-range-of-numbers-in-bash/)
[Using command line argument range in bash for loop prints brackets containing the arguments](https://stackoverflow.com/questions/5691098/using-command-line-argument-range-in-bash-for-loop-prints-brackets-containing-th)
```bash!
# This won't work
#!/bin/bash
START=1
END=5
for i in {$START..$END}
do
echo "$i"
done
# This works
#!/bin/bash
START=1
END=5
echo "Countdown"
for (( c=$START; c<=$END; c++ ))
do
echo -n "$c "
sleep 1
done
# This works
a=1
b=22
for i in $(eval echo {$a..$b});do echo $i; done
```
---
### Loop through each line of a two-column file that is delimited by space or tab. The key here is to set the internal field separator ($IFS) to $'\n' so that the for loop interates on lines rather than words. The it's simply a matter of splitting the column into individual variables. In this case, I chose to use awk since it's a simple procedure and speed is not really an issue. Long-term, I would probably re-write this using arrays.
* [Two Column for Loop in bash](https://slaptijack.com/programming/two-column-for-loop-in-bash.html)
```bash!
IFS=$'\n';
for LINE in $(cat data_file); do
VARA=$(echo ${LINE} | awk '{ print $1}')
VARB=$(echo ${LINE} | awk '{ print $2}')
echo "VARA is ${VARA}"
echo "VARB is ${VARB}"
done
```
---
### Loop thru each line of a comma- separated file, skipping 1st row (header). The difference from looping thru each line of a space or tab-separated file is that it is unnecessary to set `IFS=$'\n'`
```bash!
# Subfolders, files under $locGWAS
gwasInfoFilePath=${locGWAS}/GWAS_file_information2.csv;
# Loop thru each line of the comma-separated GWAS information file, skipping 1st row (header)
## Number of iteration: 5
count=0;
for line in `tail -n +2 $gwasInfoFilePath`;do
count=$((${count}+1));
echo "=========================================iteration $count =============================================";
dataSource=`echo $line | cut -d"," -f1`; # extract element 1 of the line
measureAbb=`echo $line | cut -d"," -f2`; # extract element 2 of the line
measureLong=`echo $line | cut -d"," -f3`; # extract element 3 of the line
folderPath=`echo $line | cut -d"," -f4 `; # extract element 4 of the line
fileName=`echo $line | cut -d"," -f5 `; # extract element 5 of the line
software=`echo $line | cut -d"," -f8` ;
filePath=`echo $line | cut -d"," -f9 `; # extract element 8 of the line
echo "line= $line";
echo "dataSource= $dataSource";
echo "measureAbb= $measureAbb";
echo "measureLong= $measureLong";
echo "folderPath= $folderPath";
echo "fileName= $fileName";
echo "software= $software";
pbs_output_dir=$folderPath/pbs_output
mkdir -p ${pbs_output_dir};
echo "qsub -N "${dataSource}_${measureAbb}" -v v_dataSource=${dataSource},v_measureAbb=${measureAbb},v_folderPath=${folderPath},v_fileName=${fileName},v_software=${software},v_filePath=${filePath} -e ${pbs_output_dir}/${dataSource}-${measureAbb}.pbs.err -o ${pbs_output_dir}/${dataSource}-${measureAbb}.pbs.out -l ncpus=1,walltime=00:30:00,mem=5gb ${jobScriptFilePath}";
qsub -N "${dataSource}_${measureAbb}" -v v_dataSource=${dataSource},v_measureAbb=${measureAbb},v_folderPath=${folderPath},v_fileName=${fileName},v_software=${software},v_filePath=${filePath} -e ${pbs_output_dir}/${dataSource}-${measureAbb}.pbs.err -o ${pbs_output_dir}/${dataSource}-${measureAbb}.pbs.out -l ncpus=1,walltime=00:30:00,mem=5gb ${jobScriptFilePath} ;
done
```
---
## which
### `which` locates a program file in the user's path. For each of its arguments which prints to stdout the full path of the executable(s). It does this by searching the directories listed in the environment variable PATH.
[Why not use “which”? What to use then?](https://unix.stackexchange.com/questions/85249/why-not-use-which-what-to-use-then)
```bash!
# Find out the path of software R
$ which R
/software/R/R-3.4.1/bin/R
```
---
## cp
### Syntax form
`cp -r /...A /...B` copies A folder and everything in it to the B folder
`cp -r /...A/ /...B` copies everything in A folder (not including A) to B folder
`cp -n A B` or `cp --no-clobber A B` A does not overwrite an existing file B [Linux how to copy but not overwrite?](https://stackoverflow.com/questions/9392735/linux-how-to-copy-but-not-overwrite)
```bash!
# Copy folder "allelicScoresCompiled" from /mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711 to /mnt/backedup/home/lunC/data/backup_PRS_UKB_201711
cp -r /mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled /mnt/backedup/home/lunC/data/backup_PRS_UKB_201711
```
---
### Copy all files from a directory to another directory
```bash!
# Copy specific files in a folder to another directory
## Copy 5 GSCAN GWAS files (*_noQIMR_noBLTS) to a LabData folder
locLabData="/mnt/backedup/home/lunC/LabData/Lab_NickM/lunC/GSCAN/data/shared_results/noQIMR_noBLTS"
mkdir -p $locLabData
cp -n $dir_destin/noQIMR_noBLTS_results/*_noQIMR_noBLTS $locLabData;
# Copy all files in a folder to another directory
dir_source="/mnt/lustre/working/lab_nickm/lunC/PRS_201802/GWASSummaryStatistics/GSCAN/noUKBiobank_results"
dir_destin2="/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/GWASSummaryStatistics/GWAS_GSCAN"
cp -n ${dir_source}/* ${dir_destin2}
```
---
## scp
### Copy a directory from one server to your server directory or vice versa
* In another account (user_B) other than yours (user_A), copy a directory to the account: `cd /user_B/destination-folder; scp -r user_A_account@user_A_hostname:/user_A/source-folder .`
* If you are on the computer wanting to receive a file from a remote computer: `scp username@remote:/file/to/send /where/to/put`
* If you are on the computer from which you want to send file to a remote computer: `scp /file/to/send username@remote:/where/to/put`
* [How to copy a file from a remote server to a local machine?](https://unix.stackexchange.com/questions/188285/how-to-copy-a-file-from-a-remote-server-to-a-local-machine)
* Copy a directory (i.e. recursively) from a remote server to another remote server: `scp -r username@hostname:/sourceDirectory /destinationDirectory`
```bash!
# Jiyuan An logged into his HPC service
cd /jiyuanAn/destination-folder
# In Jiyuan An's Shell section, copy a folder that lunC can access or own to Jiyuan An's folder
scp -r lunC@hpcpbs01.adqimr.ad.lan:/mnt/lustre/reference/data/UKBB_500k/versions/bgen201803 .
# type password by lunC
# Copy a directory /data from share.sph.umich.edu to /mnt/backedup/home/lunC/LabData-fixed/Lab_NickM/lunC/GSCAN
scp -r sftp-gscan@share.sph.umich.edu:/data /mnt/backedup/home/lunC/LabData-fixed/Lab_NickM/lunC/GSCAN/
# Type passowrd
# Download progress:
#sftp-gscan@share.sph.umich.edu's password:
#snps_associated_with_dpw.txt 100% 162KB 162.3KB/s 00:01
#noMCTFR_results.tar.gz 100% 1983MB 4.4MB/s 07:34
#no23andMe_results.tar.gz 100% 1976MB 4.3MB/s 07:44
#noUKBiobank.tar.gz 100% 1872MB 6.9MB/s 04:30
#noNTR.tar.gz 100% 1980MB 8.4MB/s 03:57
#Copy a file from a remote server to a folder in another remote server
#Copy file with updated beta (filename: no23andMe_changedBeta.tar.gz) for binary traits to a my remote folder
scp sftp-gscan@share.sph.umich.edu:/data/shared_results/no23andMe/no23andMe_changedBeta.tar.gz /mnt/backedup/home/lunC/LabData/Lab_NickM/lunC/GSCAN/data/shared_results/no23andMe/
#sftp-gscan@share.sph.umich.edu's password:
#no23andMe_changedBeta.tar.gz 88% 986MB 1.8MB/s 01:12 ETA
```
---
## basename
## dirname
### Get any part of a file path
* [Bash - get last dirname/filename in a file path argument](https://stackoverflow.com/questions/3294072/bash-get-last-dirname-filename-in-a-file-path-argument)
```bash
pathname=a/b/c
echo $(basename $pathname) # c
echo $(basename $(dirname $pathname)) # b
echo $(basename $(dirname $(dirname $pathname))) # a
```
---
## find
### list only files or subdirectorys in current directory
`find . -maxdepth 1 -type f`
`.` is the directory to search
`-maxdepth 1` limits the directory to search to 1 level (i.e., the current directory not subdirectorys)
`-type f` finds only files
`-type d` finds only directory
[List only regular files (but not directories) in current directory](https://unix.stackexchange.com/questions/48492/list-only-regular-files-but-not-directories-in-current-directory)
[Linux find folders without files but only subfolders [closed]](https://stackoverflow.com/questions/17732373/linux-find-folders-without-files-but-only-subfolders)
```bash!
# Find only files in current directory
find . -maxdepth 1 -type f > ${dir_output}/filepaths_${folder_main}.txt
# Find only directorys in current directory
find . -maxdepth 1 -type d
# .
# ./raw
# ./scRNA190620_NB551151_0102_AHT3WHBGXB_cellranger_simple
# ./scRNA190620_NB551151_0102_AHT3WHBGXB
# ./ADTB190620_NB551151_0102_AHT3WHBGXB_fixed
# ./ADTB190620_NB551151_0102_AHT3WHBGXB
# ./ADT190620_NB551151_0102_AHT3WHBGXB
```
### Find files that are non-zero in file size
* [find files with non-zero size in shell](https://serverfault.com/questions/476840/find-files-with-non-zero-size-in-shell)
```bash
# Every summedRiskProfiles.S*.log file is 0kb, as checked by the following code
find ${locArchive}/uniqSNPs_from_metaDataQCed-Release8-*AND_SNP-rsNum_from_all-QCed_GWAS-GSCAN/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-*/dosageFam_Release8_*/summedRiskProfiles.S*.log -type f -size +0
```
---
### Find absolute path of files
```bash
clumpedSNP_beta_p_UKB="${locPRSInput}/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs";
# There are 10 folders under $clumpedSNP_beta_p_UKB
ls -F $clumpedSNP_beta_p_UKB | sort
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-1000GPhase3_AND_GWAS-UKB-ASS/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-1000GPhase3_AND_GWAS-UKB-ES/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-1000GPhase3_AND_GWAS-UKB-NCD/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-1000GPhase3_AND_GWAS-UKB-NSDPW/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-1000GPhase3_AND_GWAS-UKB-SS/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ASS/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ES/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NCD/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NSDPW/
# innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-SS/
# Add absolute path of every clumpedSNPs_ALLELE1_BETA_chr* file under $clumpedSNP_beta_p_UKB to a list
find $clumpedSNP_beta_p_UKB -type f | grep clumpedSNPs_ALLELE1_BETA_chr > $clumpedSNP_beta_p_UKB/filePath_clumpedSNPs_ALLELE1_BETA # 220 files in this list (10 folders* 22 chromosomes)
```
---
### Copy or move large files using `find` command
* syntax form `find sourceFolder -name '*.*' -exec mv {} destinationFolder \;`
* [Move large number of files with find, exec mv](https://stackoverflow.com/questions/11942422/moving-large-number-of-files)
* `find sourceFolder -name '*.*'` finds the pattern specified by `-name` in `sourceFolder`
* `-exec` runs any command
* `{}` inserts the filename found
* ` \;` marks the end of the exec command
```bash!
# Example 1: copy a large number of files
## This won't work:
## This works:
find ${source_folder_path} -name "${source_file_name}" -exec cp {} ${destin_folder_path} \;
# Example 2: move a large number of files
## An error when you move a lot of files with mv
mv /mnt/lustre/working/lab_nickm/lunC/PRS_test/PRScalc/per-individualRisk* /mnt/lustre/working/lab_nickm/lunC/PRS_test/PRScalc_perIIDriskProfiles
#-bash: /usr/bin/mv: Argument list too long
# Use find and exec mv instead
locPRScalc=/mnt/lustre/working/lab_nickm/lunC/PRS_test/PRScalc
locPRScalc_perIIDriskProfiles=/mnt/lustre/working/lab_nickm/lunC/PRS_test/PRScalc_perIIDriskProfiles
find $locPRScalc -name 'per-individualRisk*' -exec mv {} $locPRScalc_perIIDriskProfiles \;
ls $locPRScalc_perIIDriskProfiles | wc -l
28260
```
---
## ls
### `ls` syntax
`ls -l` gives a long listing of all files.
`ls -r` lists the files in the reverse of the order that they would otherwise have been listed in.
`ls -t` lists the files in order of the time when they were last modified (newest first) rather than in alphabetical order.
`ls -lrt` gives a long listing, oldest first, which is handy for seeing which files in a large directory have recently been changed.
`ls -F` to add a trailing `/` to the names of directories (folders shown ended with \; files not)
```bash
$ ls -F
amino-acids.txt animals.txt elements/ morse.txt pdb/ planets.txt salmon.txt sunspot.txt
$ ls --classify
amino-acids.txt animals.txt elements/ morse.txt pdb/ planets.txt salmon.txt sunspot.txt
# list all files
ls -a
. .. amino-acids.txt animals.txt elements morse.txt pdb planets.txt salmon.txt sunspot.txt
```
---
### list all files ended with "pFinal" under a folder
```bash
locPRScalc=/working/lab_nickm/lunC/PRS_test/PRScalc
ls ${locPRScalc}/*.pFinal
/working/lab_nickm/lunC/PRS_test/PRScalc/ADHD2017.10.pFinal /working/lab_nickm/lunC/PRS_test/PRScalc/ASD2015.10.pFinal
< output skipped>
```
---
### Find folders created on a specific date and time and then delete them
```bash
cd ${locLDOutput};
# list folders under "uniqSNPs_from_metaDataQCed-Release8*/" and date time created, find those created on Feb 17 19:27, extract folder names and get their paths
ls -d -lrt uniqSNPs_from_metaDataQCed-Release8*/*/ | grep 'Feb 17 19:27' | cut -d" " -f9 | xargs realpath > folderPath_folders-created-20180217_1927-to-delete # 8 folders to delete
# delete the 8 folders
cat folderPath_folders-created-20180217_1927-to-delete | xargs rm -r
```
---
### list files with multiple patterns
* [Using OR patterns in shell wildcards](https://unix.stackexchange.com/questions/50220/using-or-patterns-in-shell-wildcards)
```bash
ls {pheno_r_CUD,pheno_r_everDrug*} > fileList_pheno_everDrug1to10AndCUD
```
---
### list folders
* `ls -F` to add a trailing `/` to the names of directories (folders shown ended with \; files not)
```bash
$ ls -F
amino-acids.txt animals.txt elements/ morse.txt pdb/ planets.txt salmon.txt sunspot.txt
$ ls --classify
amino-acids.txt animals.txt elements/ morse.txt pdb/ planets.txt salmon.txt sunspot.txt
# list all files
ls -a
. .. amino-acids.txt animals.txt elements morse.txt pdb planets.txt salmon.txt sunspot.txt
```
---
### list directory of subfolders under
```bash
# List directory of subfolders 2 level down $locASCOut/uniqSNPs_allUKBs_Release8-HRCr1.1
# -d for directory
ls -d $locASCOut/uniqSNPs_allUKBs_Release8-HRCr1.1/*/*
```
---
### Check file permission of match.pl
* -rw-r--r-- 1
* The very first character (“–” in this case) shows that the entity “match.pl” is indeed a file. If it had been a directory, we would have seen d as the very first character
* The first three (rw-) mean the owner of the file has the permission to **r**ead, **w**rite, but NOT to e**x**ecute the file
* The next three (r--) indicate that members of the same group may **r**ead the file, but cannot write or execute it
* The final three characters (r-–) mean that other users may simply read the file, but neither write nor execute it.
[file permission](https://www.linux.com/learn/understanding-linux-file-permissions)
```bash
# check file permission
[lunC@hpcpbs01 clumping]$ ls -l /mnt/lustre/home/lunC/scripts/match.pl
-rw-r--r-- 1 lunC sec_NickM 3219 Jun 12 15:39 /mnt/lustre/home/lunC/scripts/match.pl
# u: you, g: group, o:others. Here the owner can read, write and execute the file
[lunC@hpcpbs01 clumping]$ chmod u+rwx /mnt/lustre/home/lunC/scripts/match.pl
[lunC@hpcpbs01 clumping]$ ls -l /mnt/lustre/home/lunC/scripts/match.pl
-rwxr--r-- 1 lunC sec_NickM 3219 Jun 12 15:39 /mnt/lustre/home/lunC/scripts/match.pl
```
---
## du
### Summarize the fize of a folder with du
```bash
du -sh /mnt/lustre/working/lab_nickm/lunC
1.7T /mnt/lustre/working/lab_nickm/lunC
```
---
## realpath
### Get absolute path of folders
* [Getting the full path name of a given directory](https://unix.stackexchange.com/questions/258549/getting-the-full-path-name-of-a-given-directory)
```bash
clumpedSNP_beta_p_GSCAN="${locPRSInput}/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-GSCANGWASs";
# There are 10 folders under $clumpedSNP_beta_p_GSCAN. Save paths of these folders in a file
realpath $clumpedSNP_beta_p_GSCAN/* > $clumpedSNP_beta_p_GSCAN/folderPath_folder-here;
```
---
## mv
### Move a file to current location
* . means here
```bash
# current location
pwd
/mnt/backedup/home/lunC/workshop/20180206_intro_linux_python/shell-novice-data/data-shell
# Move a file to here
mv thesis/draftRenamed.txt .
```
---
## rm
### To remove same file name in multiple directories, list them first to ensure correct ones will be deleted
```bash
# Suppose there 2 folders under $locPRSoutput/uniqSNPs_allUKBs_Release8-HRCr1.1
# Each of them has 16 subfolders that contain a file fileList_withinFolder_per-individualRisk.txt to delete
locPRSoutput=/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoring/output;
# List these files
ls $locPRSoutput/*/*/*/fileList_withinFolder_per-individualRisk.txt
# Then delete them. Don't just delete them without listing them
rm $locPRSoutput/*/*/*/fileList_withinFolder_per-individualRisk.txt
# Another example
ls $locASCOut/uniqSNPs_allUKBs_Release8-HRCr1.1/*/*/PRS_UKBAllPheno*
rm $locASCOut/uniqSNPs_allUKBs_Release8-HRCr1.1/*/*/PRS_UKBAllPheno*
```
---
### Move every files except for one, a technique to archive all existing files in a folder
```bash
# Archive old files to folder $locArchive, moving everything in the folder $locLDInput to $locArchive, except for the archive folder
locLDInput="$locLDClumping/input";
locArchive="$locLDInput/archive_files_before_20180511";
mkdir -p $locArchive;
cd $locLDInput;
mv !(archive) ${locArchive}/ ;
ls
archive archive_files_before_20180511
```
---
### Move all files in a directory to another directory
`mv /path/sourcefolder/* /path/destinationfolder/`
`rm -r /path/sourcefolder/` `-r` means recursively, meaning everything in that directory
```bash
# Example 1: Archive old files under $locCommonSNPs to the archive folder under $locCommonSNPs
cd $locCommonSNPs;
mv * archive/
# Example 2: Move all files in a directory to another directory and then remove the old directory
mv /mnt/lustre/working/lab_nickm/lunC/PRS/GWAS_results/* /mnt/lustre/working/lab_nickm/lunC/PRS_test/GWAS_results
rm -r /mnt/lustre/working/lab_nickm/lunC/PRS
```
---
## xargs
### Copy multiple files with `xargs` and `cp`
```bash!
# This won't work, because as the cp error indicates, the target directory must come last.
realpath ${source_file_name_prefix}*${source_file_name_suffix} | xargs cp -n ${destin_folder_path}
#cp: target ‘/mnt/lustre/reference/data/UKBB_500k/versions/lab_stuartma/draft_gwas/BOLT_LMM/UKB-estimated-caffeine-consumed-per-day-thru-regular-coffee-and-tea_IID-NA-in-UKB20453-everUsedCannabis/BOLT-LMM-phenotype-pheno-caffeine.per.day/revised_bolt_imputed_ukb_imp_chr9_v3_caffeine.per.day.bgen.assoc’ is not a directory
```
---
### `xargs` collects the input from the pipe and then executes its arguments with the input appended. xargs takes the stdout of commandA as the parameter for commandB
`commandA | xargs commandB`
```bash!
# Get absolute paths of s*_noUKBioBank_changedBeta.txt.gz files and then decompress each of them
realpath ${dir_destin}/noUKBiobank_changedBeta/s*_noUKBioBank_changedBeta.txt.gz | xargs gunzip
```
---
### Concatenate all files which names of these files are in a separate file
* script file: `/mnt/backedup/home/lunC/scripts/PRS_UKB_201711_step01_obtainGWASSummaryStatisticsFromDiscoverySamples.sh`
```bash!
#File1 with a header row
cat test1
a b c d
1 2 3 4
#File2 with a header row
cat test2
a b c d
1 10 100 1000
#Names of file1 fiel2 in a separate file
cat fileNames
test1
test2
#Concatenate file1 and file2 excluding header rows
##awk built in variable FNR: Number of Records relative to the current input file
cat fileNames | xargs awk '(FNR>1){print $0}'> test12
cat test12
1 2 3 4
1 10 100 1000
#Write header back to the file using in-place computing
sed -i '1ia b c d' test12
cat test12
a b c d
1 2 3 4
1 10 100 1000
# Count rows in file1 and file2
cat fileNames | xargs wc -l
2 test1
2 test2
4 total
```
---
## dos2unix
### End of line (EOL) character in Windows
**Problem**: For some reason, when I open files from a unix server on my windows machine, they occasionally have Macintosh EOL conversion, and when I edit/save them again they don't work properly on the unix server. I only use notepad ++ to edit files from this unix server, so is there a way to create a macro that automatically converts EOL to Unix format whenever I open a file?
**Solution?**: Your issue may be with whatever FTP program you are using. For example, I use WinSCP to remote into a Unix server, Notepad++ is set as my default editor, but I had to go into WinSCP's settings and set the transfer mode to Binary in order to keep line endings preserved. So, you may be able to reconfigure your FTP/SCP/etc program to transfer the files in a different manner
**Solution**: That functionality is already built into Notepad++. From the "Edit" menu, select "EOL Conversion" -> "UNIX/OSX Format".

* [EOL conversion in notepad ++](https://stackoverflow.com/questions/16239551/eol-conversion-in-notepad)
---
### `dos2unix` converts DOS/MAC to UNIX text file format. Do the format conversion when an error occurs but you just cannot fine the bug. Script files created in Windows by Notepad++ or Sublime text can have Windows characters. These can cause errors when running the script file. Here is a sign of hidden Windows character.
* [dos2unix](https://www.tutorialspoint.com/unix_commands/dos2unix.htm)
```bash
# Example 1: error due to Windows format
-bash: /var/spool/PBS/mom_priv/jobs/4833550.hpcpbs02.SC: /bin/bash^M: bad interpreter: No such file or directory
# Example 2: error due to Windows format
-bash: /mnt/backedup/home/lunC/scripts/MR_ICC_GSCAN_201806/MR_step08-01-03_jobSubmit_munge-GWAS_calculate-SNP-heritability.sh: line 60: syntax error near unexpected token `$'do\r''
```
#### Remove windows character \r
```bash=
# Notice \r at the end of first line
od -c pheno_fNum_fNam
0000000 6 9 r _ n u m _ C U D \n 7 0
0000020 r _ n u m _ e v e r D r u g 1 \n
0000040 7 1 r _ n u m _ e v e r D r u
0000060 g 1 0 \n 7 2 r _ n u m _ e v e
0000100 r D r u g 2 \n 7 3 r _ n u m _
0000120 e v e r D r u g 3 \n 7 4 r _ n
0000140 u m _ e v e r D r u g 4 \n 7 5
0000160 r _ n u m _ e v e r D r u g 5 \n
0000200 7 6 r _ n u m _ e v e r D r u
0000220 g 6 \n 7 7 r _ n u m _ e v e r
0000240 D r u g 7 \n 7 8 r _ n u m _ e
0000260 v e r D r u g 8 \n 7 9 r _ n u
0000300 m _ e v e r D r u g 9 \r \n
0000315
# Change file to unix format
dos2unix pheno_fNum_fNam
# Check delimiters again. \r is gone
od -c pheno_fNum_fNam
0000000 6 9 r _ n u m _ C U D \n 7 0
0000020 r _ n u m _ e v e r D r u g 1 \n
0000040 7 1 r _ n u m _ e v e r D r u
0000060 g 1 0 \n 7 2 r _ n u m _ e v e
0000100 r D r u g 2 \n 7 3 r _ n u m _
0000120 e v e r D r u g 3 \n 7 4 r _ n
0000140 u m _ e v e r D r u g 4 \n 7 5
0000160 r _ n u m _ e v e r D r u g 5 \n
0000200 7 6 r _ n u m _ e v e r D r u
0000220 g 6 \n 7 7 r _ n u m _ e v e r
0000240 D r u g 7 \n 7 8 r _ n u m _ e
0000260 v e r D r u g 8 \n 7 9 r _ n u
0000300 m _ e v e r D r u g 9 \n
0000314
```
---
### Copying code from notepad++ in Windows 7 resulted in an error
```bash=
locPRScalc="/working/lab_nickm/lunC/PRS_test/PRScalc"
locLDcheck="${locPRScalc}/LDcheck"
declare -A pValueThresholds
pValueThresholds=( ["S1"]="0.00000005" ["S2"]="0.00001" ["S3"]="0.001" ["S4"]="0.01" ["S5"]="0.05" ["S6"]="0.1" ["S7"]="0.5" ["S8"]="1" )
# loop thru each p value threshold (S1-S8)
for pRange in {1..8};
do threshold=${pValueThresholds[S${pRange}]};
echo ${threshold};
for phenotype in ADHD2017 ASD2015;
do
echo $phenotype
for chromosome in {1..22};
do
echo "subsetting SNPs from file ${phenotype}.${chromosome}.p";
awk -v pThreshold=${threshold} '{if($2 <= pThreshold && $1 !="") print $1}' ${locPRScalc}/${phenotype}.${chromosome}.p >> ${locLDcheck}/${phenotype}.S${pRange}.snpList.txt;
done;
done;
done
# multiple errors
> g SNPs from file ${phenotype}.${chromosome}.p";
> -bash: syntax error near unexpected token `g'
> [lunC@hpcpbs01 PRScalc]$ awk -v pThreshold=${threshold} '{if($2 <= pThreshold && $1 !="") print $1}' ${locPRScalc}/${phenotype}.${chromosome}.p >> ${locLDcheck}/${phenotype}.S${pRange}.snpList.txt;
> awk: fatal: cannot open file `/working/lab_nickm/lunC/PRS_test/PRScalc/..p' for reading (No such file or directory)
> [lunC@hpcpbs01 PRScalc]$ done;
-bash: syntax error near unexpected token `done'
```
---
### Save the code in a file and convert it to unix format
* [PRS_LDcheck.sh](https://drive.google.com/open?id=0B0rTuebw8wohTE10ZXgxaEdyeDA)
```bash=
dos2unix PRS_LDcheck.sh
> dos2unix: converting file PRS_LDcheck.sh to Unix format ...
ls -l PRS_LDcheck.sh
-rw-r--r-- 1 lunC sec_NickM 808 Sep 7 11:36 PRS_LDcheck.sh
chmod +x PRS_LDcheck.sh
ls -l PRS_LDcheck.sh
-rwxr-xr-x 1 lunC sec_NickM 808 Sep 7 11:36 PRS_LDcheck.sh
# run the sh file
bash PRS_LDcheck.sh
```
---
## grep
### Search words that begin with a pattern
* see [Use grep to search for words beginning with letter “s”](https://stackoverflow.com/questions/22520324/use-grep-to-search-for-words-beginning-with-letter-s)
```bash!
# grep words that start with UKB, leaving first line out
## \b : regex anchor that means word break is not in extended regular expressions standard
sh $locScripts/fileChecker.sh $phenoRaw | grep -E '\bUKB' > qcovar_fNum_fNam
# Search words begin with pattern A (UKB) or pattern B (age) or pattern C(sex) or pattern D (PC)
sh $locScripts/fileChecker.sh $phenoRaw | grep -E '\bUKB|\bage|\bsex|\bPC'
```
---
### To search multiple patterns with a logical AND, use grep + pipe or awk + multiple patterns
* `awk '{if ($1 ~ /pattern1/ && $1 ~ /pattern2/) print $0}' file`
* `grep -E 'pattern1' file | grep -E 'pattern2'`
```bash=
# Search multiple patterns with logical ANDs using awk
$ awk '{if ($1 ~ /innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS/ && $1 ~ /dosageFam_Release8_HRCr1.1/) print $0}' ${locASCOut}/filePath_summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ASS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ES/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NCD/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NSDPW/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-SS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
# Search multiple patterns using grep
$ cat ${locASCOut}/filePath_summedRiskProfiles.S1-S8_newHeader | grep -E 'innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS' | grep -E 'dosageFam_Release8_HRCr1.1'
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ASS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-ES/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NCD/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-NSDPW/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
/mnt/lustre/working/lab_nickm/lunC/PRS_UKB_201711/allelicScoresCompiled/output/uniqSNPs_from_metaDataQCed-Release8-HRCr1.1_AND_SNP-rsNum-from-all-QCed-UKBGWASs/innerJoinedSNPsByCHRBP_metaDataQCed-Release8-HRCr1.1_AND_GWAS-UKB-SS/dosageFam_Release8_HRCr1.1/summedRiskProfiles.S1-S8_newHeader
```
---
### `grep` finds a matched pattern. `grep -v` excludes a matched pattern (i.e. invert match)
*[Negative matching using grep (match lines that do not contain foo)](https://stackoverflow.com/questions/3548453/negative-matching-using-grep-match-lines-that-do-not-contain-foo)
```bash!
# Store munged exposuure GWAS file paths in a file. grep -v excludes the outcome GWAS file.
realpath ${locLDSC_munged}/*headed.munged.sumstats.gz | grep -v 'Cannabis_ICC_UKB_small.txt.munged.sumstats.gz' > ${locLDSC_munged}/filePath_munged_exposure_GWAS_files
```
---
### Search multiple patterns with logical OR in a column.
```bash!
# Search suffix D, I, P
grep -E 'D|I|P' file
```
---
## od
[od](https://www.computerhope.com/unix/od.htm) dumps files in octal and other formats
### od syntax
`od [OPTION]... [FILE]...`
* Options
| Option | |
| -------- | -------- |
| -t TYPE, --format=TYPE | select output format or formats |
* Format Specifications
| format | |
| -------- | -------- |
| -c (same as -t c) | select ASCII characters or backslash escapes as format |
### Find delimiters with `od -t`
```bash!
# Create a file for testing od command
[lunC@hpcpbs01 ~]$ cat << __EOT__> ${locPRScalc}/pvalue.ranges.txt
S1 0.00 0.00000005
S2 0.00 0.00001
S3 0.00 0.001
S4 0.00 0.01
S5 0.00 0.05
S6 0.00 0.1
S7 0.00 0.5
S8 0.00 1.0
__EOT__
# Display the content
[lunC@hpcpbs01 ~]$ cat ${locPRScalc}/pvalue.ranges.txt
S1 0.00 0.00000005
S2 0.00 0.00001
S3 0.00 0.001
S4 0.00 0.01
S5 0.00 0.05
S6 0.00 0.1
S7 0.00 0.5
S8 0.00 1.0
# Display the contents of pvalue.ranges.txt in ASCII (character) format. As you see, all the delimiters are white spaces
[lunC@hpcpbs01 ~]$ od -t c ${locPRScalc}/pvalue.ranges.txt
[lunC@hpcpbs01 ~]$ od -c ${locPRScalc}/pvalue.ranges.txt
0000000 S 1 0 . 0 0 0 . 0 0 0 0 0 0
0000020 0 5 \n S 2 0 . 0 0 0 . 0 0 0
0000040 0 1 \n S 3 0 . 0 0 0 . 0 0 1
0000060 \n S 4 0 . 0 0 0 . 0 1 \n S 5
0000100 0 . 0 0 0 . 0 5 \n S 6 0 .
0000120 0 0 0 . 1 \n S 7 0 . 0 0 0
0000140 . 5 \n S 8 0 . 0 0 1 . 0 \n
0000157
```
---
## mkdir
### Makes directory
`rmdir directoryname` removes the directory but only if it's empty
`rm -r directoryname` removes the directory whether it's empty
* -r directory
* -f force
[`mkdir -p`](https://stackoverflow.com/questions/22737933/mkdirs-p-option) the command will create all the directories necessaries to fulfill your request, not returning any error in case that directory exists
* -p, --parents
no error if existing, make parent directories as needed
```bash!
$mkdir hello/goodbye
mkdir:cannot create directory 'hello/goodbye': No such file or directory
$mkdir -p hello/goodbye
```
---
## crontab
### Task sheduling with `crontab`
Linux Crontab Syntax. Linux crontab has six fields. 1-5 fields defines the date and time of execution. The 6’th fields are used for command or script to be executed.The Linux crontab syntax are as following. Note (1) you can use either multiple values OR a range, not a mixture, (2) Cron doesn't support fractions in the time [Crontab in Linux with 20 Useful Examples to Schedule Jobs](https://staff.washington.edu/rells/R110/)
`[Minute] [hour] [Day_of_the_Month] [Month_of_the_Year] [Day_of_the_Week] [command]`
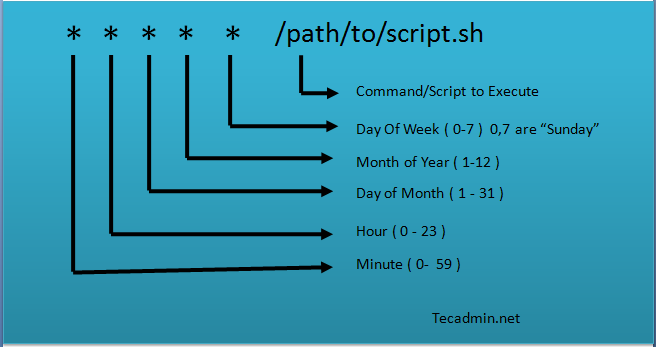
* [How to create cron statement to run for multiple hours](https://stackoverflow.com/questions/10811241/how-to-create-cron-statement-to-run-for-multiple-hours)
* [Every 7.5 minutes with cron](https://stackoverflow.com/questions/31375195/every-7-5-minutes-with-cron)
```bash!
# Wrong crontab fields 1-5
0 1-8,10-15 * * * command
# Correct crontab fields 1-5
0 1,2,3,4,5,6,7,8,10,11,12,13,14,15 * * * command
```
---
### Run a R script at regular interval using `crontab`
* [第十五章、例行性工作排程(crontab)](http://linux.vbird.org/linux_basic/0430cron.php#cron)
* [Schedule a Rscript crontab everyminute](https://stackoverflow.com/questions/38778732/schedule-a-rscript-crontab-everyminute)
```bash=
# Content of a shell file that runs a R script
cat /mnt/backedup/home/lunC/scripts/test/web-scraping_step-02_run-R-script-as-cron-jobs.sh
#!/bin/bash
# Set up directory
locScripts="/mnt/backedup/home/lunC/scripts/test"
RScriptFileName="web-scraping_step-01_jobScript_scrape-exchange-rate-AUD-to-TWD_from_DBS-bank-Taiwan.R"
RScriptFilePath=${locScripts}/${RScriptFileName}
# Load software R in order to run a R file through the Rscript command
module load R/3.4.1
# Run a R script using Rscript command
## ${RScriptFilePath} : path of the R script file to run
## arguments that will be passed into the R script file: ${trait1_name} ~ ${iteration}
Rscript --vanilla ${RScriptFilePath}
```
```shell=
# Run a R script every 5 minutes from 10 A.M. to 10 P.M. everyday
## Create a new cron
crontab -e
## To enter insert mode in Vi, type "i"
## Add a cron job
*/5 10-22 * * * /mnt/backedup/home/lunC/scripts/test/web-scraping_step-02_run-R-script-as-cron-jobs.sh
## Press Esc, then type ":wq" to save the file
## View the newly added cron job
crontab -l
*/5 10-22 * * * /mnt/backedup/home/lunC/scripts/test/web-scraping_step-02_run-R-script-as-cron-jobs.sh
## Check that you are really running crontab deamon. You should get a number as return, which is the process id for crontab.
pgrep cron
2457
## Make sure your bash file and R file are execuable
chmod 744 /mnt/backedup/home/lunC/scripts/test/web-scraping_step-02_run-R-script-as-cron-jobs.sh
chmod 744 /mnt/backedup/home/lunC/scripts/test/web-scraping_step-01_jobScript_scrape-exchange-rate-AUD-to-TWD_from_DBS-bank-Taiwan.R
```
```bash!
# Commented out cron jobs that are not working. Leave cron jobs that are working
crontab -l
#*/5 10-22 * * * /mnt/backedup/home/lunC/scripts/test/web-scraping_step-02_run-R-script-as-cron-jobs.sh
#*/5 10-22 * * * cd /mnt/backedup/home/lunC/scripts/test/ && module load R/3.4.1 && Rscript --vanilla web-scraping_step-01_jobScript_scrape-exchange-rate-AUD-to-TWD_from_DBS-bank-Taiwan.R
#* * * * * cd /mnt/backedup/home/lunC/scripts/test/ && module load R/3.4.1 && Rscript --vanilla web-scraping_step-00_jobScript_test-crontab.R
#* * * * * Rscript --vanilla /mnt/backedup/home/lunC/scripts/test/web-scraping_step-00_jobScript_test-crontab.R
#* * * * * Rscript /mnt/backedup/home/lunC/scripts/test/web-scraping_step-00_jobScript_test-crontab.R
#* * * * * /software/R/R-3.4.1/bin/Rscript /mnt/backedup/home/lunC/scripts/test/web-scraping_step-00_jobScript_test-crontab.R#`:wq
#* * * * * /software/R/R-3.4.1/bin/Rscript --vanilla /mnt/backedup/home/lunC/scripts/test/web-scraping_step-00_jobScript_test-crontab.R
*/5 10-22 * * * /software/R/R-3.4.1/bin/Rscript --vanilla /mnt/backedup/home/lunC/scripts/test/web-scraping_step-01_jobScript_scrape-exchange-rate-AUD-to-TWD_from_DBS-bank-Taiwan.R
```
---
## tar
### tar options
* `-f, --file=ARCHIVE` use archive file or device ARCHIVE
* `-v, --verbose` verbosely list files processed
* `-x, --extract, --get` extract files from an archive
* `-z, --gzip` filter the archive through gzip
### How to unpack a tar.gz file?
* [How to unpack (ungzip, unarchive) a tar.gz file](http://www.rebol.com/docs/unpack-tar-gz.html)
```bash!
cd /mnt/backedup/home/lunC/LabData/Lab_NickM/lunC/GSCAN/shared_results/noUKBiobank;
# Run the `file`command on the file to see if it says (1) gzip compressed data, last modified:... or (2) POSIX tar archive (GNU). If (1), it is correct. If (2), it means it has been uncompressed and so use `tar xf` without the `z`.
file1="/mnt/backedup/home/lunC/LabData-fixed/Lab_NickM/lunC/GSCAN/shared_results/noUKBiobank/noUKBiobank.tar.gz"
file $file1
#/mnt/backedup/home/lunC/LabData-fixed/Lab_NickM/lunC/GSCAN/shared_results/noUKBiobank/noUKBiobank.tar.gz: gzip compressed data, from Unix, last modified: Sat Feb 3 17:32:55 2018
## Unpack the file noUKBiobank.tar.gz
tar -xzf noUKBiobank.tar.gz # The result is a new directory noUKBiobank_results
```
### Untar a .tar file, extracting the files to another directory
```bash=
## Untar .tar files and extract the files to another directory ${locDownload}
tar -xvf ${locDownload}/eur_w_ld_chr.tar -C ${locDownload}
## Result: a new folder eur_w_ld_chr is created under ${locDownload} with extracted files
```
---
## bzip2
### Decompress a .bz2 file
```bash=
bzip2 -dk ${locDownload}/w_hm3.snplist.bz2 # This created a file w_hm3.snplist
```
---
## gunzip
#### Decompress A *.gz file Using The "gunzip" Command. Whilst using the "gzip" command is perfectly valid it is much easier to remember just to use `gunzip` to decompress a file as shown in the following example:
```bash!
## Decompress a .gz file
gunzip myfilename.gz
## How To Keep Both The Compressed And Decompressed File. You will now be left with "myfile" and "myfile.gz".
gunzip -k myfile.gz
```
---
## wget
### Download multiple files simultaneously
[Parallel wget in Bash [duplicate]](https://stackoverflow.com/questions/7577615/parallel-wget-in-bash)
```bash!
# Working directory
dir_working="/mnt/lustre/working/lab_marks/lunC"
dir_ENA="${dir_working}/European-Nucleotide-Archive"
dir_PRJEB25780="${dir_ENA}/PRJEB25780"
dir_download_fastq_ftp="${dir_PRJEB25780}/download_fastq_ftp"
# Change current directory to the output folder
cd ${dir_download_fastq_ftp}
# Download files in current directory without having to specify output file paths
## xargs -n 1: cat will pass one argument at a time (-n 1) to wget
## xargs -P 8 wget: execute at most 8 parallel wget processes at a time (-P 8)
cat ${dir_PRJEB25780}/fastq_ftp_URLs_no-header.tsv | xargs -n 1 -P 8 wget --limit-rate=2m
```
---
### Download a file to current directory
```bash!
cd ${dir_download_fastq_ftp}
wget --limit-rate=2m ftp.sra.ebi.ac.uk/vol1/fastq/ERR249/002/ERR2497932/ERR2497932_1.fastq.gz
```
---
### Download single .gz file
```bash!
dir_working="/mnt/lustre/working/lab_marks/lunC"
dir_data=${dir_working}/BGIvsIllumina_scRNASeq-master/data
# Download single .fastq.gz files from https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-9125/files/
mkdir -p ${dir_data}
wget -O ${dir_data}/C0088_I1.fastq.gz https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125/C0088_I1.fastq.gz
```
---
### Download all .gz files
```bash!
# Download all .fastq.gz files from https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-9125/files/
cd ${dir_data}
wget -nd -r -l1 -A gz https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125
# FINISHED --2020-10-01 14:45:40--
# Total wall clock time: 2d 4h 26m 16s
# Downloaded: 42 files, 99G in 2d 4h 23m 52s (548 KB/s)
```
---
### List URLs of all .gz files (not working)
```bash!
wget -r --spider -l2 -A gz -e robots=off https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125 2>&1
# Spider mode enabled. Check if remote file exists.
# --2020-09-29 11:02:01-- https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125
# Resolving www.ebi.ac.uk (www.ebi.ac.uk)... 193.62.193.80
# Connecting to www.ebi.ac.uk (www.ebi.ac.uk)|193.62.193.80|:443... connected.
# HTTP request sent, awaiting response... 302 Found
# Location: https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125/ [following]
# Spider mode enabled. Check if remote file exists.
# --2020-09-29 11:02:02-- https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125/
# Connecting to www.ebi.ac.uk (www.ebi.ac.uk)|193.62.193.80|:443... connected.
# HTTP request sent, awaiting response... 400 Bad Request
# Remote file does not exist -- broken link!!!
# Found 1 broken link.
# https://www.ebi.ac.uk/arrayexpress/files/E-MTAB-9125/
```
Looking at the source for the page, the href values in the actual links are things like:
```htmlmixed=
<a href="/arrayexpress/files/E-MTAB-9125/C0088_I1.fastq.gz" title="Click to download C0088_I1.fastq.gz" class="icon icon-functional" data-icon="=">
```
which are relative paths to the host website, not absolute URLs. I don't know if `wget --spider` is sophisticated enough to actually convert those to links that you can download.
Conrad
At this point I'd be looking at something like Python's excellent beautifulsoup library to read the page and construct the links you want manually.
---
## ulimit
The `ulimit` command sets or reports user process resource limits. The default limits are defined and applied when a new user is added to the system. Limits are categorized as either soft or hard. With the ulimit command, you can change your soft limits for the current shell environment, up to the maximum set by the hard limits. You must have root user authority to change resource hard limits [HOWTO: Use ulimit command to set soft limits](https://www.osc.edu/resources/getting_started/howto/howto_use_ulimit_command_to_set_soft_limits).
```bash!
ulimit -a
# core file size (blocks, -c) 0
# data seg size (kbytes, -d) unlimited
# scheduling priority (-e) 0
# file size (blocks, -f) unlimited
# pending signals (-i) 256467
# max locked memory (kbytes, -l) 64
# max memory size (kbytes, -m) unlimited
# open files (-n) 1024
# pipe size (512 bytes, -p) 8
# POSIX message queues (bytes, -q) 819200
# real-time priority (-r) 0
# stack size (kbytes, -s) 8192
# cpu time (seconds, -t) unlimited
# max user processes (-u) 4096
# virtual memory (kbytes, -v) unlimited
# file locks (-x) unlimited
# Find out the maximal user processes in the system. Each core that Cell Ranger uses will spawn 64 user processes, so use no more than 4096/64 cores
ulimit -u
#4096
```
Each core that Cell Ranger uses will spawn 64 user processes, so you may run into problems if your system has a limit on the max user processes, 4069. You can use `ulimit -u` to find out the limit.