# NESI Workflow Seminar
## Usefull resources
- [Melbourne tutorial](https://www.melbournebioinformatics.org.au/tutorials/tutorials/cwl/media/#1)
- [eResearch slides](https://jdeligt.github.io/presentations/2020_eResearch_BOF_Workflows.html#1)
- [protocol](https://link.springer.com/protocol/10.1007/978-1-4939-9074-0_24)
---
I'll go through the slidedeck but here's the gist of it:

*source [xkcd.com]*
---
# So what can we do to help our future self?
---
## code DRY (“Don’t Repeat Yourself”)
*we'll leave the wet stuff for in the lab*
A valuable lesson from the [how not to code](https://blog.usejournal.com/how-to-write-bad-code-the-definitive-guide-part-i-15b944e4cd3e) guidebook.
---
In your own code this means embracing functions:

*source [https://learncodingwithgo.com/go-functions/]*
In pipelines it means embracing workflow languages:

*source [https://bcbio-nextgen.readthedocs.io/en/latest/contents/cwl.html]*
---
## It's about defining what needs to happen and having tools or pipelines that can do that and can easily be swapped for other/newer tools.
---
Queue [eResearch slides](https://jdeligt.github.io/presentations/2020_eResearch_BOF_Workflows.html#1)
---
# Demo time
---
### Prep I did
```bash
cd /scale_wlg_persistent/filesets/project/nesi02659/workflow_workshop/
# Data
wget https://www.melbournebioinformatics.org.au/tutorials/tutorials/cwl/media/data/data.tar.gz
tar -zxvf data.tar.gz
# Tool definitions
git clone https://github.com/common-workflow-language/workflows
# A genomics workflow
git clone https://github.com/EvolutionaryGenomics/scalability-reproducibility-chapter.git
```
---
Prep NeSI environment
```bash
module load cwltool/3.0.20200317203547-gimkl-2020a-Python-3.8.2
module load Clustal-Omega/1.2.2-gimkl-2017a
module load snakemake/5.10.0-gimkl-2020a-Python-3.8.2
```
---
## Using pre defined tools
### BWA-MEM
```cwl
#!/usr/bin/env cwl-runner
cwlVersion: v1.0
class: CommandLineTool
requirements:
DockerRequirement:
dockerPull: "quay.io/biocontainers/bwa:0.7.17--ha92aebf_3"
inputs:
InputFile:
type: File[]
format: http://edamontology.org/format_1930 # FASTA
inputBinding:
position: 201
Index:
type: File
inputBinding:
position: 200
secondaryFiles:
- .fai
- .amb
- .ann
- .bwt
- .pac
- .sa
#Optional arguments
Threads:
type: int?
inputBinding:
prefix: "-t"
MinSeedLen:
type: int?
inputBinding:
prefix: "-k"
...
```
```bash
# Running it
cwltool --singularity workflows/tools/bwa-mem.cwl \
--reads core/mutant_R1.fastq \
--reads core/mutant_R2.fastq \
--reference core/wildtype.fna \
--output_filename mutant.sam
```
Output
```bash
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 24960 sequences (3744000 bp)...
[M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (0, 12080, 0, 0)
[M::mem_pestat] skip orientation FF as there are not enough pairs
[M::mem_pestat] analyzing insert size distribution for orientation FR...
[M::mem_pestat] (25, 50, 75) percentile: (372, 398, 425)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (266, 531)
[M::mem_pestat] mean and std.dev: (398.41, 39.71)
[M::mem_pestat] low and high boundaries for proper pairs: (213, 584)
[M::mem_pestat] skip orientation RF as there are not enough pairs
[M::mem_pestat] skip orientation RR as there are not enough pairs
[M::mem_process_seqs] Processed 24960 reads in 1.281 CPU sec, 1.281 real sec
[main] Version: 0.7.12-r1039
[main] CMD: bwa mem /var/lib/cwl/stgeafa3970-3893-4462-80a8-b3d02c449898/wildtype.fna /var/lib/cwl/stg69a63b06-1720-4bb9-8860-33393993ad0d/mutant_R1.fastq /var/lib/cwl/stg65485d7b-a5db-475e-8335-1ebcbe42cb76/mutant_R2.fastq
[main] Real time: 1.447 sec; CPU: 1.355 sec
INFO [job bwa-mem.cwl] completed success
{
"output": {
"location": "file:///scale_wlg_persistent/filesets/project/nesi02659/workflow_workshop/mutant.bam",
"basename": "mutant.bam",
"class": "File",
"checksum": "sha1$ea94193808e79eb3eb296b7fd3280652104db8cc",
"size": 10035037,
"path": "/scale_wlg_persistent/filesets/project/nesi02659/workflow_workshop/mutant.bam"
}
}
```
---
## Using a pre defined workflow
[source](https://github.com/EvolutionaryGenomics/scalability-reproducibility-chapter/blob/master/Snakemake/Snakefile)
```python
TARGETS = list(map(lambda n: "../data/cluster%05d/results0-3.txt" % n, range(1, 73)))
rule all:
input:
expand("{cwd}/{target}", cwd=os.getcwd(), target=TARGETS)
rule clustal:
input:
"{cluster}/aa.fa"
output:
guidetree = "{cluster}/aa.ph",
align = "{cluster}/aa.aln"
shell:
"clustalo -i {input} --guidetree-out={output.guidetree} > {output.align}"
rule pal2nal:
input:
"{cluster}/aa.aln"
output:
"{cluster}/alignment.phy"
shell:
"pal2nal.pl -output paml {input} {wildcards.cluster}/nt.fa > {output}"
rule codeml:
input: "{cluster}/alignment.phy"
output: "{cluster}/results0-3.txt"
shell:
"cd {wildcards.cluster}; echo | codeml ../paml0-3.ctl"
```
### Running it snakemake
```bash
cd scalability-reproducibility-chapter/Snakemake
snakemake
```
### Running it CWL
```bash
cd scalability-reproducibility-chapter/
cwltool --singularity CWL/workflow.cwl --clusters data
```
-----
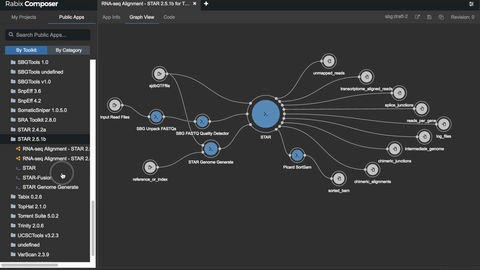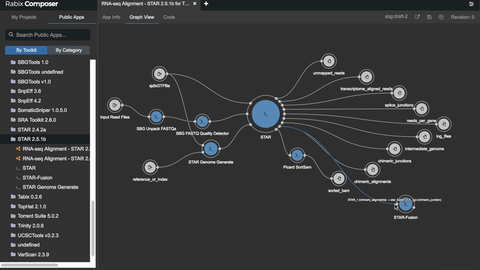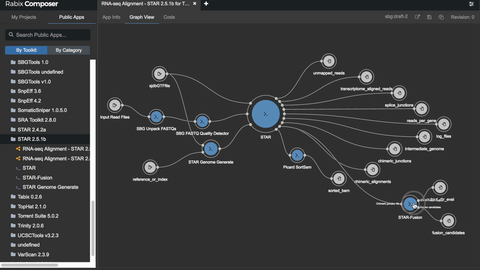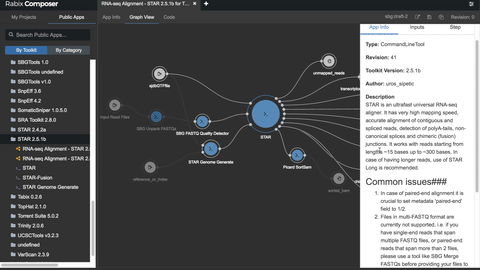
# Unlocking the potential
- [bio-tools](https://github.com/common-workflow-library/bio-cwl-tools)
- [workflow viewer](https://view.commonwl.org/workflows)
- [human genomics specific workflows](https://github.com/genome/analysis-workflows)
- many many more
### Standing on the shoulders of really tall people & awesome groups
*source SevenBridges*