Broad Institute GATK === ###### tags: `基因體/二級分析` ###### tags: `生物資訊`, `基因體`, `二級分析`, `GATK` <br> [TOC] <br> > https://software.broadinstitute.org/gatk/ <br /> ## 基因體定序 導讀 - [WGS (Whole Genome Sequencing,全基因體定序)](https://hackmd.io/qmzwm4idRyWROeeboqpfSw) <br /> ## GATK 導讀 - **全名**: - Genome Analysis Toolkit (基因體分析工具包) - **開發者**: - Broad Institute (博勞德研究所) - MAP: https://goo.gl/maps/ArufnvfyvTb8DMD16 - 隸屬於麻省理工學院和哈佛大學 - **開發目的**: - 分析人類的「基因組」和「外顯子」,找出 [SNPs](https://unclegene6666.pixnet.net/blog/post/308333779) 和 [INDELs](https://www.scmh.org.tw/internet/blog/Blog_Preview.aspx?Blog=Q0FfREVQVDAwMQ==&Article=48807515-ca94-47b6-8f85-5832b9b78d5e) - 案例:[對世界各地高粱品種的 SNP 研究](http://toolsbiotech.blog.fc2.com/blog-entry-49.html) - **核心功能**: - 用來分析基因體,尋找變異的地方 - 晚期延伸應用: - 分析 RNA 序列 - 應用到其他物種 - **版本演進**: - 早期: - [單一核苷酸多型性 (Single Nucleotide Polymorphisms, SNPs)](https://unclegene6666.pixnet.net/blog/post/308333779) - [插入與缺失 (Insertions and Deletions, INDELs)](https://www.scmh.org.tw/internet/blog/Blog_Preview.aspx?Blog=Q0FfREVQVDAwMQ==&Article=48807515-ca94-47b6-8f85-5832b9b78d5e) - 晚期加入了: - 拷貝數變異 (Copy Number Variations, CNVs) - 染色體結構變異 (Structure Variations, SVs) - [短變異檢測(short variant calling)](https://software.broadinstitute.org/gatk/documentation/tooldocs/current/) - **Picard 工具**: - 功能: - 處理高通量定序資料 - SAM / BAM / CRAM / VCF 的格式轉換 - ```$ ./gatk --list```,後面有標注 (Picard) 皆是,非常多 - 開發語言: - Java - [2017 年 6 月釋出 GATK4,並進行開源](https://kknews.cc/zh-tw/tech/68l8pzp.html) > 基於新架構開發,大幅精簡了獨立工具,並對性能增強技術(如Apache SparkTM) 提供了支援。新框架帶來了平行計算上的提升,以雲部署為依託,讓大容量基因資料分析比以往更便捷、更快速、更高效。 > 「這是基因組科學的一大步,它的性能提升與可擴展性為生物技術、製藥等所有研究領域提供了機會。在英特爾的新參考架構下,GATK4將達到五倍於早期版本的速度。」 > ——Jason Waxman,英特爾數據中心解決方案部副總裁兼總經理 - **工具說明:** - **指令**: - 一種類似 git 命令列操作模式的指令 - [GATK4 命令列語法](https://software.broadinstitute.org/gatk/documentation/article?id=11050) - [GATK4 工具清單](https://software.broadinstitute.org/gatk/documentation/tooldocs/current/)  - **開發語言**: - 核心:Java (目前使用 1.8) - 腳本:python (用於打包 java 指令) - 其他:R 語言 - **支援平台**:  - 支援 Linux 和 MacOS X,不支援 Windows - 備註: - 有點說不過去,Windows 皆可執行上述語言 - (但 Windows XP 無法安裝 Java 8) - 應該是在於:腳本有用到 shell script 撰寫(sh file)? - **安裝流程**: - 下載 GATK 並解壓縮 https://software.broadinstitute.org/gatk/download/ - 最基本的指令測試 ```$ ./gatk``` <br /> ## Genome Analysis Toolkit (基因體分析工具包) - Variant Discovery in High-Throughput Sequencing Data 探索在高通量定序資料中的變異  - Developed in the Data Sciences Platform at the Broad Institute, the toolkit offers a wide variety of tools with a primary focus on variant discovery and genotyping. Its powerful processing engine and high-performance computing features make it capable of taking on projects of any size. 該工具包由 Broad Institute (MIT 博德研究所) 的資料科學平台開發,提供各種工具,主要關注在「變異發現」和「基因分型」。 其強大的處理引擎和高效能計算功能,使其能夠承擔任何規模的專案。 (on-going) <br /> ## GATK 標準分析流程 - 又稱 GATK Best Practise (GATK [典範實務](http://mypaper.pchome.com.tw/angelo_chen/post/1283275875)) - 底下(有不同來源的)三張圖,描述 GATK 三大處理階段/程序: 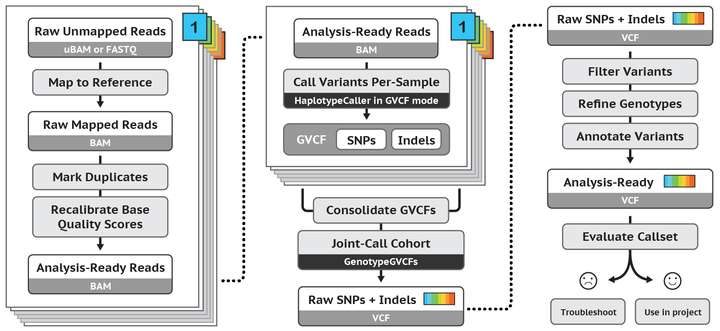 (圖片來源:https://zhuanlan.zhihu.com/p/33515262) - 資料前處理 - 尋找變異點 - 資料解讀 <br />  (圖片來源:官網 https://software.broadinstitute.org/gatk/documentation/article?id=3238) <br /> 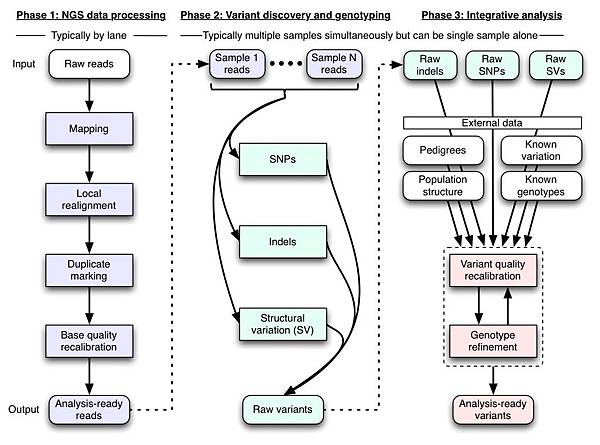 (圖片來源:https://yourgene.pixnet.net/blog/post/94090920) ### 階段一 > 次世代定序 資料處理 (NGS data processing)/資料清理(data cleanup) - 原始定序片段 (Raw reads) - 比對 (Mapping) - 局部重新比對 (Local realignment) - 重複標示 (Duplicate marking) - 序列品質再校正 (Base quality recalibration) - 準備好要分析的定序片段 (Analysis-ready reads) ### 階段二 > 變異探索與基因分型 (Variant discovery and genotyping) ### 階段三 > 整合分析 (Integrative analysis) (on-going) <br> ## GATK 使用的方法與演算法 > https://software.broadinstitute.org/gatk/documentation/topic?name=methods - [Best Practices for Variant Discovery in DNAseq](https://software.broadinstitute.org/gatk/documentation/article?id=3238) - [Best Practices for Variant Discovery in RNAseq](https://software.broadinstitute.org/gatk/documentation/article?id=4067) - [HC overview: How the HaplotypeCaller works](https://software.broadinstitute.org/gatk/documentation/article?id=4148) (on-going) <br> ## 檔案格式(file format) ### bed file format (.bed 檔) - 功能: - 用來描述「鹼基序列」被標注的資訊 - 有點像是在玩拼圖的時候,直接參照全圖來找位置 - 格式: - 每一列為一筆資訊,有3個必要欄位、9個選擇性欄位 - **欄位1**:chrom - 定義:染色體(chromosome)或支架(scaffold)的名稱  ([圖片來源與說明](http://blog.sina.com.cn/s/blog_a893ace40101hum2.html)) config:片段重疊群, scaffold:支架 (可視為小群組&大群組) - **欄位2**:chromStart - 定義:標準染色體座標中,特徵的起始位置 - base = 0 - [chromStart = 2 表示第 3 個鹼基](https://blog.csdn.net/sunchengquan/article/details/85019083) - **欄位3**:chromEnd - 定義:標準染色體座標中,特徵的結束位置(不含) - [chromEnd = 5 表示第 6 個鹼基,其特徵不含此鹼基](https://blog.csdn.net/sunchengquan/article/details/85019083) - 資料順序: - 根據「欄位1」排序 - 若「欄位1」相同,則根據「欄位2」排序 - > The intervals MUST be sorted by coordinate (in increasing order) within contigs; and the contigs must be sorted in the same order as in the sequence dictionary. This is require for efficiency reasons. - **參考資料**: - [BED File Format - Definition and supported options](https://asia.ensembl.org/info/website/upload/bed.html) - [bed文件格式详解](https://blog.csdn.net/herokoking/article/details/79276513) <br> ### [VCF (Variant Call Format) (.vcf 檔)](https://hackmd.io/6rATKTvURVSKia8K_9kBeQ) <br> ## 參考資料 - 官網(Broad Institute) - [gatk 首頁](https://software.broadinstitute.org/gatk/) - [gatk 命令列語法](https://software.broadinstitute.org/gatk/documentation/article?id=11050) - [初识 GATK](https://zhuanlan.zhihu.com/p/33515262) - 有勁基因 - [有勁基因 GATK ( Genome Analysis Toolkit)](https://yourgene.pixnet.net/blog/post/94090920) - [生物資訊工具介紹--- VarDict](https://yourgene.pixnet.net/blog/post/116518362) - 遗传病测序数据分析解读科普系列 - [第3.1期 变异发现细节:走进HaplotypeCaller内部](https://bbs.genelinks.com/thread-1076-1-1.html) <br> ## 附錄 - [xxx.vcf](https://hackmd.io/JhM5KGz7Q9yIzZ6_eXQxDA)
×
Sign in
Email
Password
Forgot password
or
Sign in via Google
Sign in via Facebook
Sign in via X(Twitter)
Sign in via GitHub
Sign in via Dropbox
Sign in with Wallet
Wallet (
)
Connect another wallet
Continue with a different method
New to HackMD?
Sign up
By signing in, you agree to our
terms of service
.