<!-- .slide: data-background="https://raw.githubusercontent.com/maxulysse/maxulysse.github.io/main/assets/img/svg/green_white_bg.svg" -->
<a href="https://www.nf-co.re"><img src="https://raw.githubusercontent.com/nf-core/logos/master/byte-size-logos/bytesize-darkbg.svg" width="65%"><img></a>
# \#22: nf-core/eager
James A. Fellows Yates / <img src="https://openmoji.org/data/color/svg/E040.svg" width=50> @jfy133 / <img src="https://openmoji.org/data/color/svg/E045.svg" width=50> @jfy133
_Hans Knöll Institute / Max Planck Institute for Evolutionary Anthropology_
---
# Overview
> nf-core/eager is a bioinformatics best-practice analysis pipeline for NGS sequencing based **ancient DNA (aDNA)** data analysis.
- What is **palaeo**genomics
- And why (do) we need a special pipeline
- Overview of nf-core/eager pipeline
- Development challenges during DSL1
---
# What is palaeogenomics?
- **Palaeogenomics** research is diverse:
- 👶 genomes for studying human history
- 🦣 genomes for past ecology/evolution
- 🦠 genomes for studying past disease
- 🧫 microbiomes for past disease, human behaviour
- :national_park: sediment DNA for ecology/evolution, human history
---
# Overview of EAGER (v1)
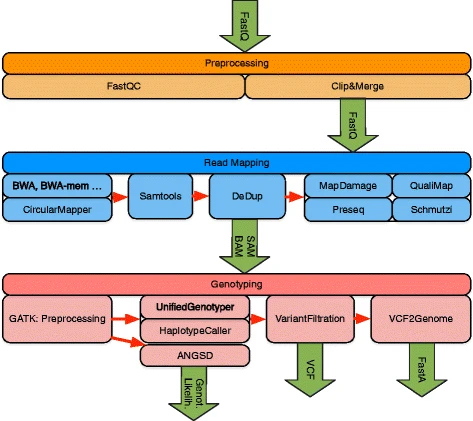
[_Peltzer et al. 2016 Genome Biology_](https://doi.org/10.1186/s13059-016-0918-z)
---
# Isn't that just genomics?
- Preprocessing :arrow_right: mapping :arrow_right: genotyping
- Pretty standard, no?
- Except, _ancient_ DNA (aDNA) is shitty
- Fragmented
- Damaged
- Mostly (modern) contamination
- Complicates things...
---
# What does that mean?

<aside class="notes">
- Fragmentation -> Low alignment specificity -> Short sequences: can map to many places
- Too much fragmentation -> Fragments lost -> Low coverage: low variant calling confidence
- Still OK, but less confidence? -> Short AND _damaged_ (artificial deamination changes C to Ts at ends of reads) -> Complicates variant calling/Complicates taxonomic profiling
- Further complicated! -> contamination -> Cross-mapping from environmental relatives -> which is right call?
</aside>
---
# Not all is lost
- 😢 Shitty DNA makes things difficult
- 💡 Helps to distinguish between aDNA and modern DNA
- Authentication criteria:
- ✅ Damage profiles
- ✅ Fragments length distributions
- ✅ Edit distances
- ✅ Metagenomic component like modern samples?
---
# Scaling palaeogenomics
- 👨🏿🔬 Nowadays: **easy** to get aDNA
- 📈 Problem: too good, 1000s of samples!
- Previous pipelines not for HPCs
- 👭 **Interdisciplinary** analyses more common
- e.g. Combine human pop-gen with pathogen detection
---
# Solution
<img src="https://i.imgur.com/tbWUnQS.png" width="50%">
---
# nf-core/eager

<aside class="notes">
So what are we doing to adapt to aDNA?
- Relaxing mapping parameters (more mismatches)
- Generating damageprofilers
- Clipping off damage
- Filtering for just damage reads
- Estimating nuclear contamination (human)
- Estimating edit distances
</aside>
---
# nf-core/eager
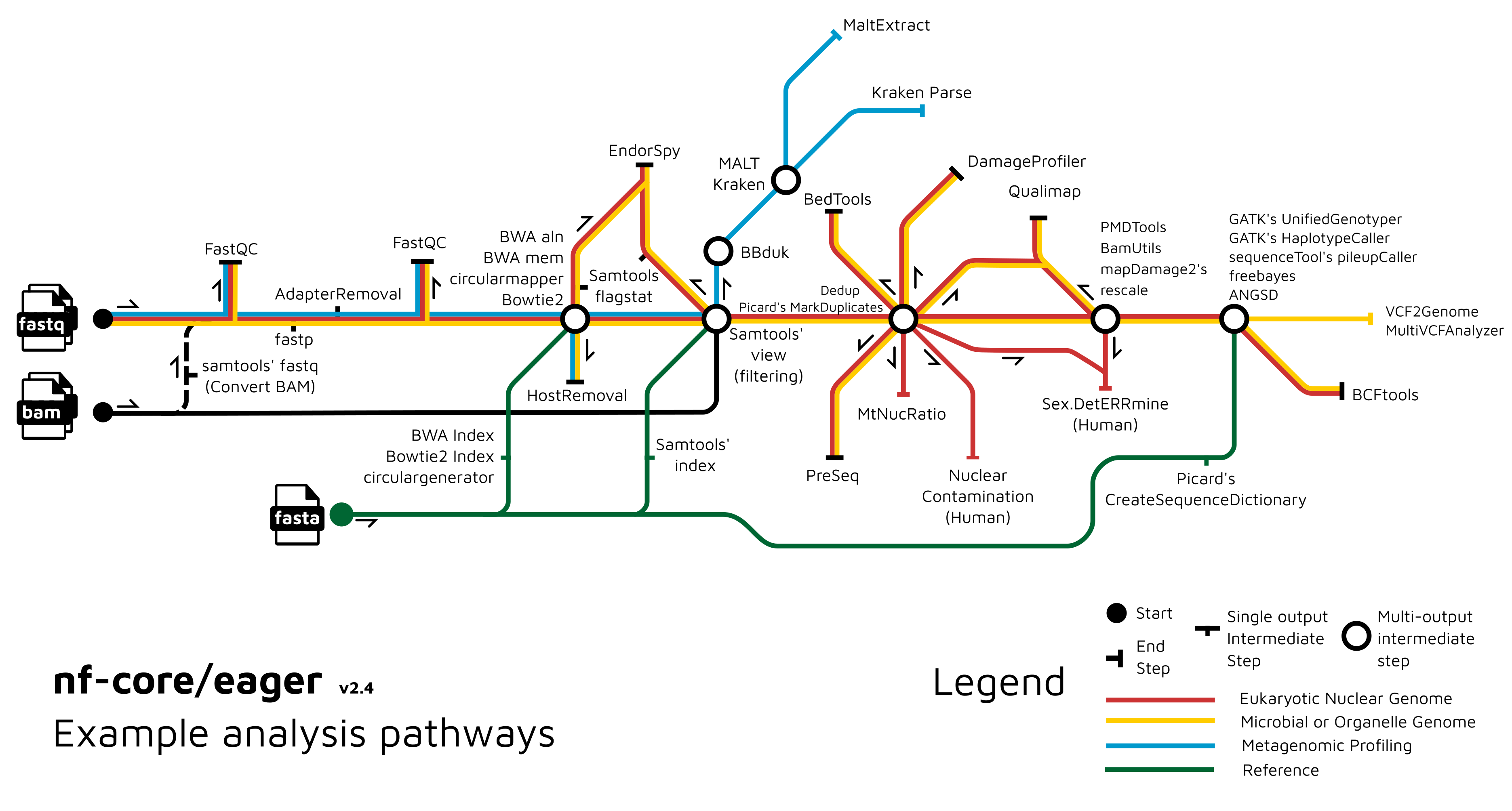
---
# Main Development Challenges
---
# Issue: complex input data :spaghetti:
- Different library treatments
- No, half, or full damage removal
- Mix many different sequencing configs
- paired and single end
- MiSeq/NextSeq/HiSeq/NovaSeq/BGI
- Heterogeneous input files
- Start with FASTQ, sometimes BAM
- Already adapter clipped, sometimes not
---
# Solution: TSV input and 'rerouting'
**Lots** of channel branching, filtering etc.
```groovy=
if (params.complexity_filter_poly_g) {
ch_input_for_fastp = ch_convertbam_for_fastp.branch{
twocol: it[3] == '2' // Nextseq/Novaseq data with possible sequencing artefact
fourcol: it[3] == '4' // HiSeq/MiSeq data where polyGs would be true
}
} else {
ch_input_for_fastp = ch_convertbam_for_fastp.branch{
twocol: it[3] == "dummy" // seq/Novaseq data with possible sequencing artefact
fourcol: it[3] == '4' || it[3] == '2' // HiSeq/MiSeq data where polyGs would be true
}
}
<...>
ch_skipfastp_for_merge.mix(ch_fastp_for_merge)
.into { ch_fastp_for_adapterremoval; ch_fastp_for_skipadapterremoval }
```
---
# Issue: very broad user base 👩🏿💻
- Bioinformaticians
- Genomicists
- Ecologists
- Archaeologists
- Osteologists
- Historians
- Amateur genealogists
- ...
---
# Solution: Docs, docs, docs, docs...
- ...docs, docs docs
- So much Phil & co. complained 😉
- Complex pipeline: How to keep 'interesting'?
- Descriptive images and schematics!
- Write for re-use as broad training material!
- Educated students _before_ starting project
<img src="https://raw.githubusercontent.com/nf-core/eager/2.4.0/docs/images/output/fastqc/fastqc_adapter_content.png" width="45%">
---
# Issue: Lots of opinions, no standards :speaking_head_in_silhouette:
- Young and very competitive field
- Human population genetics in particular!
- Constantly changing 'standards' (i.e. strong opinions)
- Difficult to know what tool or parameter to use...
🤦♀
---
# Solution: be open and pester
- Develop tool ecosystem
- Twitter hivemind is your friend
- Repeatedly present in different contexts
- Offer workshops!
<img src="https://i.imgur.com/89sZ7yU.png" width=30%>
<img src="https://i.imgur.com/ny9Pe0B.png" width=30%>
<img src="https://i.imgur.com/BkAzrlb.png" width=30%>
---
# Summary
- 🦣 Palaeogenomics is complicated
- Topic variety, shitty DNA, complicated processing
- But fun challenge!
- 📚 Broad documentation helps in interdisciplinary fields
- 📣 Be active in outreach (not just support!)
- Helps keep a project alive past publication
---
## Need help?
<!-- .slide: data-background="https://raw.githubusercontent.com/maxulysse/maxulysse.github.io/main/assets/img/svg/green_white_bg.svg" -->
Repository: [`nf-core/eager`](https://github.com/nf-core/eager)
Tutorials: [`https://nf-co.re/eager/usage#tutorials`](https://nf-co.re/eager/usage#tutorials)
Chat: [`https://nf-co.re/join`](https://nf-co.re/join) <img src="https://cdn.brandfolder.io/5H442O3W/at/pl546j-7le8zk-6gwiyo/Slack_Mark.svg" width=5%></img>`#eager`
Publication: [`10.7717/peerj.10947`](https://doi.org/10.7717/peerj.10947)
<b>Thanks</b>: Alex, co-devs, bug reporters, testers etc.!
<p align="center">
Follow nf-core on
<a href="https://www.twitter.com/nf_core"><img src="https://openmoji.org/data/color/svg/E040.svg" width=6%></a>
<a href="https://github.com/nf-core"><img src="https://openmoji.org/data/color/svg/E045.svg" width=6%></a>
<a href="https://www.youtube.com/c/nf-core"><img src="https://openmoji.org/data/color/svg/E044.svg" width=6%></a>
</a>
</p>
<div style="display: flex; justify-content: space-evenly; align-items:center;">
<img src="https://chanzuckerberg.com/wp-content/themes/czi/img/logo.svg" width=15%>
<a href="https://nf-co.re/" style="color: #000000; font-family:Monaco, monospace; font-weight:bold;font-size:18pt">https://nf-co.re/</a>
<div style="font-style:italic; font-size: 0.5em; color: #000000;">Icons:<br><a href="https://openmoji.org">openmoji.org</a></div></div>
<style>
.reveal section img { background:none; border:none; box-shadow:none; }
body {
background-image: url(https://raw.githubusercontent.com/nf-core/logos/master/nf-core-logos/nf-core-logo-square.svg);
background-size: 7.5%;
background-repeat: no-repeat;
background-position: 3% 96%;
background-color: #181a1b;
}
.reveal body {
font-family: 'Roboto', sans-serif;
font-weight: 300;
color: white;
}
.reveal p {
font-family: 'Roboto', sans-serif;
font-weight: 300;
color: white;
}
.reveal h1 {
font-family: 'Roboto', sans-serif;
font-style: bold;
font-weight: 400;
color: white;
font-size: 62px;
}
.reveal h2 {
font-family: 'Roboto', sans-serif;
font-weight: 300;
color: white;
}
.reveal h3 {
font-family: 'Roboto', sans-serif;
font-style: italic;
font-weight: 300;
color: white;
}
.reveal p {
font-family: 'Roboto', sans-serif;
font-weight: 300;
color: white;
}
.reveal li {
font-family: 'Roboto', sans-serif;
font-weight: 300;
color: white;
}
.reveal pre {
background-color: #272822 !important;
display: inline-block;
border-radius: 7px;
color: #aaaba9;
}
.reveal pre code {
color: #eeeeee;
background-color: #272822;
font-size: 100%;
}
.reveal code {
background-color: #272822;
font-size: 75%;
}
.reveal .progress {
color: #24B064;
}
.reveal .controls button {
color: #24B064;
}
.reveal blockquote {
display: block;
position: relative;
width: 90%;
margin: 20px auto;
padding: 5px;
background: rgba(255, 255, 255, 0.05);
box-shadow: 0px 0px 2px rgb(0 0 0 / 20%);
}
</style>
{"metaMigratedAt":"2023-06-16T11:05:01.175Z","metaMigratedFrom":"YAML","title":"nf-core/bytesize 22 eager","breaks":true,"contributors":"[{\"id\":\"6357fb8d-b0f8-4b7d-9fbe-6d813ad67700\",\"add\":18712,\"del\":13792}]"}