---
title: 'Converting PGX pipeline to Hydra'
disqus: hackmd
tags: Projects
---
Converting PGX pipeline to Hydra
===



## Table of Contents
[TOC]
## Hydra aln module DAG

## Make the environment
This should be done only the first time!
```Python=
sudo -i
mamba env create -f /home/massi/Documents/GMS/hydra_genetics/hydra_test/hydra_0.15.0/env.yml
# if you want to change any of the package's version:
mamba env update -f=/home/massi/Documents/GMS/hydra_genetics/hydra_test/hydra_0.15.0/env.yml --prune
# if you want to search for the latest version of a package:
mamba search -c bioconda snakefmt
```
This is the env.yml file used with hydra 0.2.0:
```bash
name: hydra-genetics_0.15.0
channels:
- conda-forge
- bioconda
- defaults
dependencies:
- pandas[version='>=1.3.1']
- snakemake==7.14.0
- singularity==3.8.5
- jinja2==3.0.3
- snakefmt=0.6.1
- pycodestyle=2.8.0
- pytest=7.1.2
- networkx
- spyder=5.3.0
- spyder-unittest=0.5.0
# - click==8.0
# Type checking tool setup
- mypy = 0.950 # type checker
- pytest = 7.1.2 # testing framework
- pytest-mypy = 0.8.0 # pytest plugin
- pytest-cov = 3.0.0
# - black = 22.3.0
- flake8 = 4.0.1
- pylint = 2.13.9
- yapf = 0.32.0
- pip>=22.0
- pip:
- hydra-genetics==0.15.0
```
## Clone the repository
Exit the sudo mode and activate the new environment to test hydra installation:
```bash=
clone https://github.com/Genomic-Medicine-Linkoping/pgx.git
cd ~/Documents/GMS/hydra_genetics/hydra_test/hydra_0.15.0/pgx
conda env list
conda activate hydra-genetics_0.15.0
hydra-genetics --help
```
You should get this screen:
```bash
_ _ _ _ ____ ____ __ ___ ____ __ _ ____ ____ __ ___ ____
/ )( \( \/ )( \( _ \ / _\ ___ / __)( __)( ( \( __)(_ _)( )/ __)/ ___)
) __ ( ) / ) D ( ) // \(___)( (_ \ ) _) / / ) _) )( )(( (__ \___ \
\_)(_/(__/ (____/(__\_)\_/\_/ \___/(____)\_)__)(____) (__) (__)\___)(____/
hydra-core/tools version 0.15.0
Usage: hydra-genetics [OPTIONS] COMMAND [ARGS]...
CLI tool to prepare and initialize snakemake projects
Options:
-v, --verbose Print verbose output to the console.
-l, --log-file <filename> Save a verbose log to a file.
--help Show this message and exit.
Commands:
create-input-files create input-files, samples.tsv and units.tsv
create-module create bare bone project
create-rule add rule to project
referece-data download reference data
```
## Create a new rule
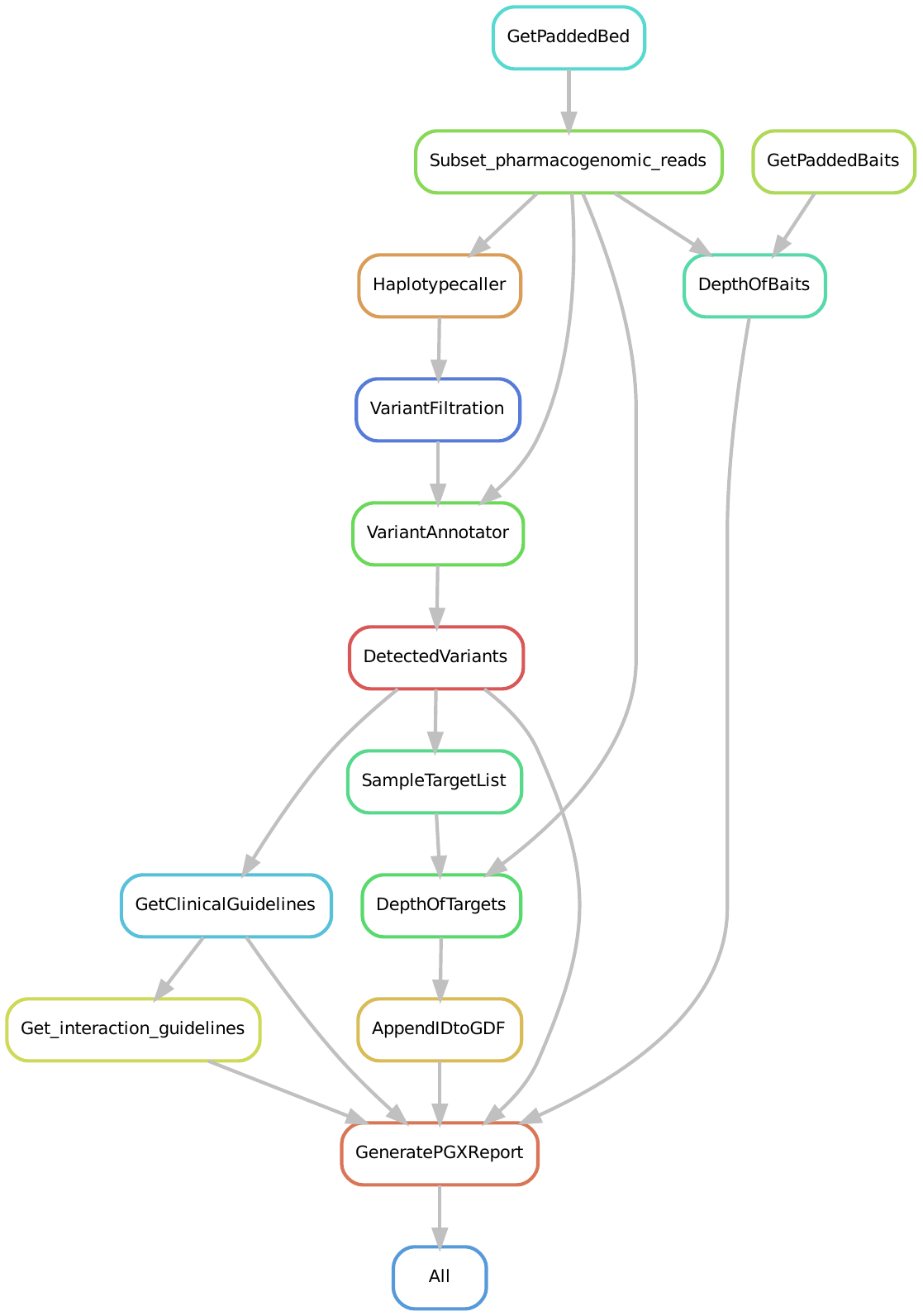
Lauri converted the first rule HaplotypeCaller so we want to swith to that branch to create a new one from wich we want to convert the next rule:
```bash=
git switch migrate_rule_HaplotypeCaller
# create the new branch and switch it
git switch -c migrate_rule_VariantFiltration
hydra-genetics create-rule --command variant_filtration --module pgx --author "Lauri Mesilaakso & Massimiliano Volpe" --email "Lauri.Mesilaakso@regionostergotland.se & massimiliano.volpe@liu.se"
# it will ask the command name, we typed Python but we modified all the filenames later on
more ./workflow/rules/python.smk
```
or if they fix something in Uppsala you better switch to develop after pulling those changes:
```bash=
git switch develop
# create the new branch and switch it
git switch -c migrate_rule_DetectedVariants
git push --set-upstream origin migrate_rule_DetectedVariants
hydra-genetics create-rule --command detected_variants --module pgx --author "Massimiliano Volpe" --email "massimiliano.volpe@liu.se"
# it will ask the command name, we typed Python but we modified all the filenames later on
more ./workflow/rules/python.smk
```
After that make sure those files are on point:
```bash=
config/config.yaml
envs/rule_name.yaml
rules/rule_name.smk
rules/common.smk
scripts/script_name.py
.tests/integration/config.yaml
```
## Run the module
With Singularity support:
```bash
snakemake --cores 12 \
--use-singularity \
--configfile config/config.yaml \
--singularity-args "--bind /mnt/WD1/ref" \
--forceall \
-s workflow/Snakefile
```
With Conda support:
```bash
snakemake --cores 12 \
--configfile config/config.yaml \
--use-conda \
--forceall \
-s workflow/Snakefile
```
The new rule will be added to the job list:
```python
Building DAG of jobs...
Using shell: /bin/bash
Provided cores: 12
Rules claiming more threads will be scaled down.
Conda environments: ignored
Job stats:
job count min threads max threads
-------------------------------- ------- ------------- -------------
alignment_picard_mark_duplicates 4 1 1
alignment_samtools_extract_reads 4 1 1
alignment_samtools_index 5 1 1
all 1 1 1
filtering_variant_filtration 4 1 1
padd_target_regions 1 1 1
snv_indels_bed_split 4 1 1
snv_indels_haplotypecaller 4 12 12
variant_annotator 4 1 1
total 31 1 12
Select jobs to execute...
```
## Test the module
Snakefmt provides formatting specifications for Snakemake files.
The library it's not working properly so we will use the singularity container instead:
```bash!=
#snakefmt "-l 130" --compact-diff .
mkdir tmp
singularity pull tmp/snakefmt.sif docker://quay.io/biocontainers/snakefmt:0.6.0--pyhdfd78af_0
singularity exec tmp/snakefmt.sif snakefmt "-l 130" --compact-diff .
#INFO: Converting SIF file to temporary sandbox...
#=====> Diff for workflow/rules/common.smk <=====
#=====> Diff for workflow/Snakefile <=====
#=====> Diff for workflow/rules/padd_target_regions.smk <=====
#=====> Diff for workflow/rules/variant_annotator.smk <=====
#=====> Diff for workflow/rules/filtering.smk <=====
#[INFO] All done
singularity exec tmp/snakefmt.sif snakefmt "-l 130" .
#INFO: Converting SIF file to temporary sandbox...
#[INFO] Writing formatted content to workflow/rules/variant_annotator.smk
#[INFO] All done 🎉
#INFO: Cleaning up image...
```
Linting is the automated checking of your source code for programmatic and stylistic errors:
```bash!=
snakemake --lint text --configfile config/config.yaml
# Congratulations, your workflow is in a good condition!
# on .tests/integration run this:
snakemake --lint -n -s ../../workflow/Snakefile --configfile config/config.yaml
# if you need json output:
# snakemake --lint json --configfile config/config.yaml
```
Pycodestyle is a tool to check your Python code against some of the style conventions in PEP8:
```bash!=
pycodestyle --max-line-length=130 --statistics workflow/scripts
#workflow/scripts/variant_filtration.py:12:131: E501 line too long (144 > 130 characters)
#1 E501 line too long (144 > 130 characters)
yapf --in-place --verbose workflow/scripts/*.py
```
## Commit to GitHub
```bash=
VisualStudio git section
drop-down menu
Semantic Commit
Write the message and choose the semantic feature
Click on the Commit buttton
```
## Test the module
```bash!
snakefmt "-l 130" \
--compact-diff \
.
singularity exec tmp/snakefmt.sif \
snakefmt "-l 130" \
--compact-diff \
.
singularity exec tmp/snakefmt.sif \
snakefmt "-l 130" \
.
snakemake \
--lint workflow/Snakefile filtering_variant_filtration
snakemake --lint text --configfile config/config.yaml
pycodestyle \
--max-line-length=130 \
--statistics workflow/scripts
yapf --in-place --verbose workflow/scripts/*.py
```
User story
---
```gherkin=
Feature: Guess the word
# The first example has two steps
Scenario: Maker starts a game
When the Maker starts a game
Then the Maker waits for a Breaker to join
# The second example has three steps
Scenario: Breaker joins a game
Given the Maker has started a game with the word "silky"
When the Breaker joins the Maker's game
Then the Breaker must guess a word with 5 characters
```
> I choose a lazy person to do a hard job. Because a lazy person will find an easy way to do it. [name=Bill Gates]
```gherkin=
Feature: Shopping Cart
As a Shopper
I want to put items in my shopping cart
Because I want to manage items before I check out
Scenario: User adds item to cart
Given I'm a logged-in User
When I go to the Item page
And I click "Add item to cart"
Then the quantity of items in my cart should go up
And my subtotal should increment
And the warehouse inventory should decrement
```
> Read more about Gherkin here: https://docs.cucumber.io/gherkin/reference/
User flows
---
```sequence
Alice->Bob: Hello Bob, how are you?
Note right of Bob: Bob thinks
Bob-->Alice: I am good thanks!
Note left of Alice: Alice responds
Alice->Bob: Where have you been?
```
> Read more about sequence-diagrams here: http://bramp.github.io/js-sequence-diagrams/
Project Timeline
---
```mermaid
gantt
title A Gantt Diagram
section Section
A task :a1, 2014-01-01, 30d
Another task :after a1 , 20d
section Another
Task in sec :2014-01-12 , 12d
anther task : 24d
```
> Read more about mermaid here: http://mermaid-js.github.io/mermaid/
## Appendix and FAQ
:::info
### Online resources
**Find this document incomplete?** Leave a comment!
- [Hydra-Genetics Best Practice](https://docs.google.com/document/d/1l2v1ItZBTDaI72vQPZcaQzxwVUao78XzIJvGASAAD9E/edit#)
- [Conventional commits specs](https://www.conventionalcommits.org/en/v1.0.0/)
- [Semantic Commit messages](https://gist.github.com/joshbuchea/6f47e86d2510bce28f8e7f42ae84c716)
- [semantic-git-commit-cli](https://www.npmjs.com/package/semantic-git-commit-cli) is nice but didn't manage to get it to work with singularity. I used the following VScode extension instead
- [Git - Semantic Commit](https://marketplace.visualstudio.com/items?itemName=nitayneeman.git-semantic-commit)
- [Git Squash](https://stackoverflow.com/questions/5189560/squash-my-last-x-commits-together-using-git)
:::
###### tags: `GMS` `PGX` `hydra-genetics`
 Sign in with Wallet
Connect another wallet
Sign in with Wallet
Connect another wallet