1. # Testing for (https://EG333)DNA Damage in herbarium sp[](https://)1. # Testing for (https://EG333)DNA Damage in herbarium sp[](https://)1. # Testing for (https://EG333)DNA Damage in herbarium sp[](https://)1. # Testing for (https://EG333)DNA Damage in herbarium sp[](https://)
### Ancient DNA damage
DNA damage is so widespread in ancient specimens that it is actually used as a verification step to ensure the DNA is not contaminated with modern DNA. A few statistics are collected:
- Read length: expected to be much shorter than for modern specimens
- lambda: the probability of single-stranded overhangs
- deltaS: Cytosine deamination probability in a single-stranded context
- C-to-T proportion: a "ski-slope" graph showing higher proportion of C-to-T SNPs early in reads (image from MapDamage website):
### "Antique" specimens
Do these patterns hold for more recently collected, but still old specimens? Instead of ancient-DNA, we may say these are "antique" DNA. There have only been a few investigations of this in plants:
#### [Weiss et al., 2016 Herbarium Specimen Damage](https://royalsocietypublishing.org/doi/full/10.1098/rsos.160239#d3e1152)
* Whole genome sequencing of herbarium specimens of *Arabidopsis* and *Solanum* spanning 250 years.
* Significant association between nucleotide misincorporation and specimen age

#### [Wales et al., 2017 Sunflower Domestication](https://onlinelibrary.wiley.com/doi/pdfdirect/10.1111/eva.12594)
> In addition, the δS parameter calculated by mapDamage provides a probability of cytosine deamination in single-stranded contexts (Table S1). Our samples produced δS values ranging from 0.165 to 0.999 (mean = 0.605). As anticipated from well-preserved, relatively recent specimens, the ethnographic samples exhibit low levels of damage (δS range = 0.018–0.056, mean = 0.035)
From their table S1, their most recent non-ethnographic sample (900 ybp) had a δS of 0.5 while the age of the ethnographic samples was typically 1920s - 1930s.
[Example of their "ski jump plots" from MapDamage](https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1111%2Feva.12594&file=eva12594-sup-0003-FigS3.pdf)
#### [Gutaker et al., 2019 Potato domestication](https://www.nature.com/articles/s41559-019-0921-3)
- Potato herbarium specimens from 1680 - 1900
- Using their data in Supplement, C-T probability is significantly correlated with age (r^2 = 0.37) (my plot of this below)

- [Supplementary Fig. 2](https://static-content.springer.com/esm/art%3A10.1038%2Fs41559-019-0921-3/MediaObjects/41559_2019_921_MOESM1_ESM.pdf) has MapDamage results with higher proportions of C-T SNPs (5-10%) in first 5 bp.
#### [Forrest et al., 2020 Preservation Methods and Plant DNA](https://internal-journal.frontiersin.org/articles/10.3389/fevo.2019.00439/full)
- Looked at several peparation methods for newly collected specimens, including drying with various techniques, and pickling.
- They did not see any sigifnicant signal with MapDamage
- However [they do see high C-T SNPs in pickled specimens](https://www.frontiersin.org/files/Articles/483035/fevo-07-00439-HTML-r2/image_m/fevo-07-00439-g006.jpg).
- Also looked at the effect of using [FFPE](https://www.neb.com/products/m6630-nebnext-ffpe-dna-repair-mix#Product%20Information) as a way to repair DNA before library prep; they did see a reduction in SNPs in most repaired DNA.
#### [McGaughran 2020 targeted sequencing moth museum specimens](https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6594-0)
- [Limited effect of age on C-to-T SNPs](https://media.springernature.com/lw685/springer-static/image/art%3A10.1186%2Fs12864-020-6594-0/MediaObjects/12864_2020_6594_Fig1_HTML.png?as=webp).
- Not sure what's going on with the high G-to-A rate for a set of 1970s specimens, it's not addressed in the text.
- Uracil-Specific Excision Reagent [(USER) Enzyme](https://www.neb.com/products/m5508-thermolabile-user-ii-enzyme#Product%20Information) may have contributed. This enzyme is also used in NEB Oligo kits.
## MapDamage
https://ginolhac.github.io/mapDamage/
### Installation
Create a new conda environment with only what is needed and install MapDamage via bioconda, along with other tools needed for this analysis:
`sudo conda create -n mapdamage mapdamage2 bwa samtools gatk4`
activate conda environment: `conda activate mapdamage`
command: `mapDamage`
#### Inputs
* A BAM file with a correct header
* A FASTA file with reference sequences
They recommend only including overlapping reads for highly degraded samples.
#### Outputs
* Plot with a fragmentation misincorporation patterns
* Plot showing frequencies of C -> T 5' and G -> A 3'
## The Goals
- Identify the extent of DNA damage in herbarium specimens sequenced via target capture.
- Determine the effect of not accounting for DNA damage in:
- Population genetics
- Phylogenetics
## Part 1: Mapping target capture data to a reference genome
### Data
Target capture data from *Artocarpus altilis* herbarium specimens spanning 200 years:
```csvpreview {header="true"}
Bioinformatic ID Collection Taxon Year HerbariumID
Aa_BS_Epoly Banks and Solander s.n. A. altilis 1769 V
Aa_US008_Caribbean A. Ricksecker 488 A. altilis 1896 US
Ac_US007_Caribbean Britton & Cowell 285 A. camansi 1901 US
Aa_US37_CentralAmerica Aforie 820 A. altilis 1902 US
Aa_FM2_Caribbean C.f. baber 137 A. altilis 1904 F
Ac_US017_Caribbean Merrill 830 A. camansi 1915 US
Aa_AH011_Maire_EPoly G.P. Wilder 161001 A. altilis 1926 BISH
Aa_AH006_Maohi_EPoly G.P. Wilder 160994 A. altilis 1926 BISH
Aa_AH010_Tuutou_EPoly G.P. Wilder 160952 A. altilis 1927 BISH
Aa_AH009_PaeTau_EPoly G.P. Wilder 160980 A. altilis 1927 BISH
Aa_AH014_Raumai_EPoly G.P. Wilder 160956 A. altilis 1927 BISH
Aa_AH008_Puriati_EPoly G.P. Wilder 160969 A. altilis 1927 BISH
Aa_US36_CentralAmerica Standley 53750 A. altilis 1928 US
Aa_US36_CentralAmerica Standley 53750 A. altilis 1928 US
Ac_FM7_SouthAmerica E. Matuda 1946 A. camansi 1948 F
Aa_US35_Caribbean Little 13601 A. altilis 1950 US
Aa_US35_Caribbean Little 13601 A. altilis 1950 US
Am_AH029_Micro Fosberg 34417 A. marianennsis 1952 US
Aa_FM5_CentralAmerica Antonio Molina R. 10167 A. altilis 1961 F
Am_AH027_Micro Fosberg 47441 A. marianennsis 1965 US
Aa_US34_Caribbean Reed 1999 A. altilis 1967 US
Am_AH026_Micro Fosberg 58755 A. marianennsis 1978 US
Aa_US33_Caribbean Howard 19124 A. altilis 1979 US
Aa_AH020_Caribbean Pruski 1574 A. altilis 1980 NY
Ac_FM6_SouthAmerica Leonardo Grefa EE 83 A. camansi 1985 F
Aa_DR200_Maire_EPoly D Ragone DR200 A. altilis 1987 PTBG
Aa_AH019_Caribbean Steven R. Hill 21337 A. altilis 1990 NY
Aa_US018_Caribbean E. Stijfhoorn 887 A. altilis 1992 US
Aa_US006_Africa J. Fernandez Casas A. altilis 1993 US
Aa_FM4_Mela Takeuchi & Waikabu 15185 A. altilis 1994 F
```
*Artocarpus altilis* reference genome from [Sahu et al., 2020](https://www.mdpi.com/2073-4425/11/1/27/htm).
`wget https://bioinformatics.psb.ugent.be/gdb/aocc/artal/Artal_genome_LATEST.fa.gz`
### Setup Genome and Reads
#### Genome Reference
`bwa index Artal_genome_LATEST.fa`
#### Map reads
`bwa mem altilis_genome/Artal_genome_LATEST.fa reads/A_altilis_BS_combined.R* | samtools view -bS - > alignments/A_altilis_BS.genome.bam`
Parallel version (forward read only):
`parallel "bwa mem altilis_genome/Artal_genome_LATEST.fa reads/*{}*R1*_paired.fastq | samtools view -bS > alignments/{}.R1.bam" :::: altilis_samples.txt`
Parallel version (both reads):
`parallel "bwa mem altilis_genome/Artal_genome_LATEST.fa reads/*{}*R*_paired.fastq | samtools view -bS - > alignments/{}.genome.bam" :::: altilis_samples.txt`
Sort and index:
`parallel samtools sort alignments/{}.genome.bam -o alignments/{}.sorted.bam :::: altilis_samples.txt`
`parallel samtools index alignments/{}.sorted.bam :::: altilis_samples.txt`
Mark Duplicates:
`parallel gatk MarkDuplicates -I alignments/{}.sorted.bam -O alignments/{}.marked.bam -M alignments/{}.markeddups.txt :::: altilis_samples.txt`
In the MapDamage2.0 paper (supplemental) they further process the alignment by:
Only using overlapping reads
[FilterUniqueBAM.py](https://github.com/shendurelab/cfDNA/blob/master/FilterUniqueBAM.py)
### Run MapDamage
#### Initial tests
`mapDamage -i alignments/A_altilis_BS.genome.bam -r altilis_genome/Artal_genome_LATEST.fa`
Got a warning:
```
WARNING: Alignment contains a large number of reference sequences (98152)!
This may lead to excessive memory/disk usage.
Consider using --merge-reference-sequences
```
MapDamage seems to work single-threaded.
Took about 20 minutes to run.
Re-running with the `--merge-reference-sequences option` reduces the run time to only 5 minutes.
#### Parallel commands
Initial Run:
`parallel mapDamage -i alignments/{}.marked.bam -r altilis_genome/Artal_genome_LATEST.fa --merge-reference-sequences :::: altilis_samples.txt`
Change Y axis on plots:
`parallel mapDamage -d results_{}.marked -y 0.1 --plot-only :::: altilis_samples.txt`
Rescaling:
`parallel mapDamage -d results_{}.marked --rescale-only -i alignments/{}.marked.bam -r altilis_genome/Artal_genome_LATEST.fa :::: altilis_samples.txt`
rescaling gives a warning about most of the reads, wonder if it's because of the de-duplication:
```
Warning! Assuming the pairs are non-overlapping, facing inwards and correctly paired.
Number of non-rescaled reads due to improper pairing: 511560
```
**Repeat with consensus reads?**
### Results: DNA Damage in *Artocarpus altilis* herbarium specimens
### delta-s by Year
- there is a relationship, but the 1700s sample is an outlier

- Without the 1700s specimen, r^2 for delta-s is 0.25

### percent of DNA damage by year
- The older specimens do have a bit of an uptick in C-to-T SNPs in the first few bp.
- Way smaller values than either the Weiss or Wales papers

- there is a relationship, but the 1700s sample is an outlier
- Without the 1700s specimen, r^2 for 1stbp is 0.14

### How much does it matter?
How can we get at the effect size without doing an entire masters' thesis worth of popgen?
### Fixing the oldest specimen
Try again after removing all reads > 200 bp
```
fastp --length_limit 200 -i A_altilis_BS_combined.R1.paired.fastq -o A_altilis_BSnolong_combined.R1.paired.fastq -I A_altilis_BS_combined.R2.paired.fastq -O A_altilis_BSnolong_combined.R2.paired.fastq
```
## Analysis without a reference genome
The availability of a reference genome is a major impediment to using MapDamage for most target capture data, which is done without a reference genome.
Can we use the supercontigs (exons and flanking non-coding regions) from deeply sequenced modern specimens to define a "reference genome," which can be used to compare older specimens?
This won't be a fit for all circumstances, as many taxa are used from herbarium specimens precisely because there are no modern specimens available.
However, identifying the DNA damage pattern is common to all herbarium specimens of certain age may allow us to use lab or bioinformatic methods that don't rely on MapDamage.
### Data
Pairs of Artocarpus HybSeq datasets, one recent and one preserved.
```csvpreview {header="true"}
Run Library samplename Rundir Fileprefix Species year
NA US008 US008 NA US008_S13 altilis 1896
4 EGL94 900260-001 run4 900260-001_S13 altilis 2000
3 EGL73 BB18789 run3comb EGL73comb_S17 altissimus 1934
6 EGL222 EG441 run6 EG441_S66 altissimus 2016
7 EGL242 Fuchs21347 run7 EGL242_S16 corneri 1963
6 EGL212 EG333 run6 EG333_S56 corneri 2016
7 EGL231 Dunlop5189 run7 EGL231_S5 glaucus 1979
2 EGL38 NZ852 run2 NZ852_S14 glaucus 2013
7 EGL238 Krukoff4372 run7 EGL238_S12 griffithii 1932
2 EGL50 NZ216 run2 NZ216_S26 griffithii 2002
6 EGL181 Beguin1900 run6 Beguin1900_S23 horridus 1920
6 EGL221 EG437 run6 EG437_S65 horridus 2016
3 EGL35 Taam2259 run3comb EGL35comb_S1 hypargyraeus 1941
6 EGL187 EG170 run6 EG170_S31 hypargyraeus 2016
6 EGL180 S31741 run6 S31741_S22 obtusus 1972
6 EGL203 EG248 run6 EG248_S47 obtusus 2016
7 EGL254 Elmer12468 NA EGL254_S28 treculanis 1910
2 EGL34 NZ203 NA NZ203_S10 treculanis 2000
3 EGL78 Elmer16247 run3comb EGL78comb_S22 xanthocarpus 1916
4 EGL91 Yang15648 run4 Yang15648_S10 xanthocarpus 2003
```

Need to transfer the supercontigs from the recent specimens and the trimmed reads from the older specimens.
### Setup
Extract supercontigs for each new sample:
```python=
import os,sys
from Bio import SeqIO
new_namelist = [x.rstrip() for x in open("new_namelist.txt")]
genefiles = os.listdir("big_phylogeny_supercontigs")
for newID in new_namelist:
print(newID)
genecount = 0
with open("supercontigs/{}.supercontigs.fasta".format(newID),'w') as outfile:
for gene in genefiles:
geneID = gene.replace(".supercontig.raw.fasta","")
for seq in SeqIO.parse(os.path.join("big_phylogeny_supercontigs",gene),'fasta'):
if seq.id.endswith("_{}".format(newID)):
seq.id = seq.id + "-{}".format(geneID)
seq.description = ''
SeqIO.write(seq,outfile,'fasta')
genecount+=1
print("{} genes".format(genecount))
```
Make a `species_pairs.txt` file that has the new sample, old sample, and species name:
```
EG170 EGL35 hypargyraeus
EG248 S31741 obtusus
EG333 EGL242 corneri
EG437 Beguin1900 horridus
EG441 EGL73 altissimus
NZ216 EGL238 griffithii
NZ852 EGL231 glaucus
Yang15648 EGL78 xanthocarpus
```
Bash script to contain the workflow through MapDamage:
```bash=
set -eo pipefail
new=$1
old=$2
species=$3
echo $new
echo $old $species
mkdir -p $species
cd $species
source activate mapdamage
bwa index ../supercontigs/$new.supercontigs.fasta
bwa mem ../supercontigs/$new.supercontigs.fasta ../reads/old/"$old"*.fastq | samtools view -bS - > $old.$new.bam
samtools sort $old.$new.bam -o $old.$new.sorted.bam
samtools index $old.$new.sorted.bam
gatk MarkDuplicates -R ../supercontigs/$new.supercontigs.fasta -I $old.$new.sorted.bam -M $new.dups.txt -O $old.$new.marked.bam
mapDamage -i $old.$new.marked.bam -r ../supercontigs/$new.supercontigs.fasta --merge-reference-sequences -y 0.1
```
Run on all the pairs of samples:
`parallel --colsep ' ' bash map_by_sp.sh {1} {2} {3} :::: species_pairs.txt`
### Analysis
#### Distribution of DNA damage in old/new pairs
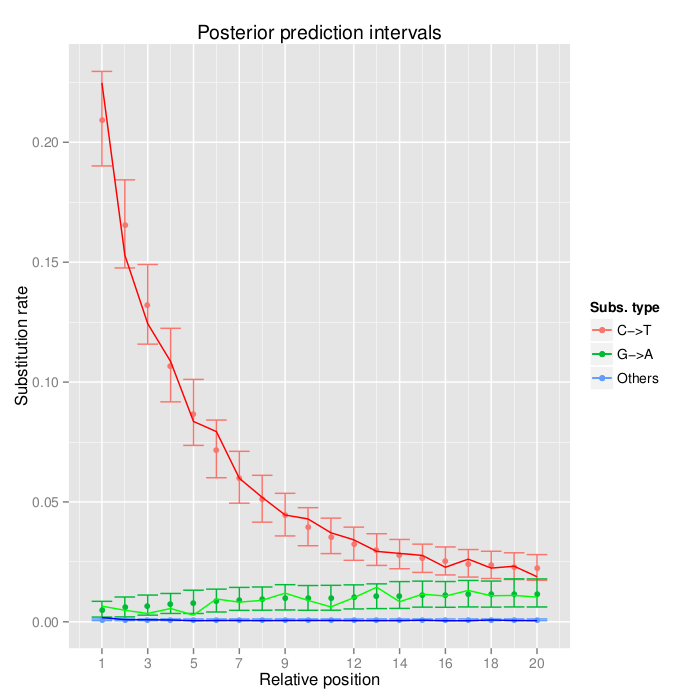
Two samples had high deltaS scores: EGL78 (*A. xanthocarpus* from 1916, deltaS = 0.53) and EGL73 (*A. altissimus* from 1934, deltaS = 0.61). Both values are much higher than anything seen with *A. altilis*.
#### Phylogeny with damaged herbarium supercontigs
Tree with no chomping - red specimens are modern, blue are herbarium.

#### Phylogeny after trimming damaged herbarium specimens more aggressively
Use FASTP to trim 5 bp from the beginning and end of every read.
```
parallel fastp -I ../to_supercontigs/reads/old/{}*R1*_paired.fastq -O reads/{}.trim5.R1.fastq -i ../to_supercontigs/reads/old/{}*R2*_paired.fastq -o reads/{}.trim5.R2.fastq -f 8 -t 8 -F 8 -T 8 -h {}.html -j {}.json :::: old_namelist.txt
```
Re-run HybPiper for these samples
`while read name; do /opt/apps/Software/HybPiper/reads_first.py -r reads/$name* -b newref.fna --bwa --prefix $name --cpu 50; python /opt/apps/Software/HybPiper/intronerate.py --prefix $name; python /opt/apps/Software/HybPiper/cleanup.py $name; done < old_namelist.txt`
MAFFT
`parallel "mafft --preservecase --localpair --maxiterate 1000 unaligned/{}.supercontig.chomped.fasta > mafft/{}.supercontig.chomped.mafft.fasta" :::: ../../trimmed/candidate_genes.txt`
TRIMAL
`parallel trimal -gt 0.5 -in mafft/{}.supercontig.chomped.mafft.fasta -out trimal/{}.supercontig.chomped.trimal.fasta :::: ../../trimmed/candidate_genes.txt`
Remove gene name from sequence name so they can be combned:
`parallel sed -i "s/-{}//g" {}.supercontig.chomped.trimal.fasta :::: ../../../trimmed/candidate_genes.txt`
Combine
`python /opt/apps/Software/HybPiper/fasta_merge.py --fastafiles trimal/*.fasta > artocarpus.chomped.concat.fasta`
Phylo
`iqtree -s artocarpus.chomped.concat.fasta -m MFP -B 100`
Repeat with original supercontigs, no chomping
`parallel "mafft --preservecase --localpair --maxiterate 1000 unaligned/{}.supercontig.untrimmed.fasta > mafft/{}.supercontig.untrimmed.mafft.fasta" :::: ../../trimmed/candidate_genes.txt`
`parallel trimal -gt 0.5 -in mafft/{}.supercontig.untrimmed.mafft.fasta -out trimal/{}.supercontig.untrimmed.trimal.fasta :::: ../.trimmed/candidate_genes.txt`
`parallel sed -i "s/-{}//g" {}.supercontig.untrimmed.trimal.fasta :::: ../../../trimmed/candidate_genes.txt`
`python /opt/apps/Software/HybPiper/fasta_merge.py --fastafiles trimal/*.fasta > artocarpus.untrimmed.concat.fasta`
#### Tree with trimmed reads
8 bp removed from either end of every read for herbarium specimens (in blue)
