###### tags: `BOT3404`
# SESSION 2: HIGH-THROUGHPUT DNA SEQUENCE FILES
[TOC]
## Bioinformatic Research in Biology
> Bioinformatics is the study of biology using the tools of computer science. Bioinformatics enables the study of proteins, genes and genomes using computer algorithms and computer databases. According to a more general National Institutes of Health definition (see PDF below), bioinformatics is "research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, organize, analyze, or visualize such data." The related discipline of computational biology is "the development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems."
SOURCE: [Kennedy Krieger Institute](https://www.kennedykrieger.org/research/centers-labs-cores/bioinformatics)
Bioinformatics is inherintly associated to biology research. However, the use of bioinformatics is not limited to the subjects covered in this tutorial. While it has prevalent use in genomics and genetics projects, computational skills utilized in this workshop can be applied to research in nearly all discliplines within biology. Not only is processing data from wet lab part of bioinformatics, but the visualization of data is a major aspect of bioinformatics work.
## Short Reads Sequencing
When we sequence short reads, we mean that the genomic code (DNA) has been fragmented into small chunks, called 'reads'. We do this because we are unable to sequence very long stretches of DNA, and could potentially lose data. In the wetlab procedure, DNA extraction materials are fragmented with an enzyme called fragmentase. The seqments are amplified using PCR, and then paired together in the library preparation step. This is why data rendered from sequencing will have two files for one sample. One is the forward read, and one is the reverse read.

SOURCE: [The Sequencing Center](https://thesequencingcenter.com/knowledge-base/what-are-paired-end-reads/)
## Opening and Viewing a FASTQ files
Last week, you calculated the number of lines and the sizes of two FASTQ files, which contain the raw output of a DNA sequencer. The sequences are _paired_ which means that the first sequence in the `R1.paired.fastq` file correpsonds to the same DNA fragment as the first sequence in the `R2.paired.fastq` file.
The files you worked with last week were already _trimmed_ meaning that poor quality DNA sequences were removed from the file. This week, you will work with _untrimmed_ files and use statistics about the sequences to decide on quality control for the sequences.
A FASTQ file is also a text format, but contains the raw or modified output of DNA sequencers, such as the Illumina system. Each sequence in a FASTQ file is known as a "read" and is represented by four lines:
1. The identifier for the read
2. The DNA sequence
3. A placeholder line, usually just a `+` character
4. [Quality scores represented by a single character.](https://en.wikipedia.org/wiki/Phred_quality_score)
There are untrimmed sequence files in `/scratch/bot3404/reads`. You will be assigned to a specific pair of files starting with `ABRONIAseq`. The files will end with `.fastq.gz` which means the files have been compressed to save space on the computer.
**Copy the two sequence files you have been assigned to your directory on SPHAGNUM**:
`cp /scratch/bot3404/reads/Abronia/ABRONIAseq999.R1.fastq.gz /scratch/bot3404/yourname/`
### `zcat`
You cannot normally use `cat` to view compressed files, (give it a try to see some crazy looking output!) but you can use `zcat` instead:
`zcat filename.fastq.gz`
**Use `zcat` to view one of your compressed `fastq.gz` files**. There will be a lot of output! You can use Control-C to cancel.
You can pipe the output of `zcat` to other programs. For example, you can get the number of lines in the file:
`zcat filename.fastq.gz | wc -l`
You can also use `less` to view the sequence files:
`zcat filename.fastq.gz | less`
**Questions**
1. How does the number of lines using `wc -l` correspond with the number of sequences in the file?
2. Are all the sequences the same length?
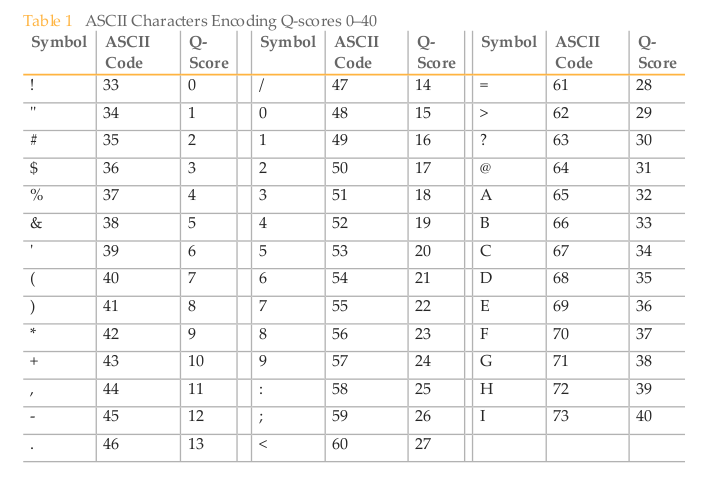
3. The image below has the conversion between ASCII characters and quality scores (Q-scores) for DNA sequences. Look through some of your sequences - are the scores high (above 30) or low (below 20)?
## Using FastQC to Evaluate Quality of Reads
FastQC is a tool used to perform quality control functions on untrimmed sequence data. It will also provide a set of summary statistics that allow us to identify problems in the data before continuing with the pipeline. FastQC creates an HTML report which can be opened outside of your terminal. You can read more about the function of FastQC on the [Babraham Bioinformatics](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) site. Here are links to [bad](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/bad_sequence_fastqc.html) and [good](https://www.bioinformatics.babraham.ac.uk/projects/fastqc/good_sequence_short_fastqc.html) outputs.
FastQC accepts multiple input fastp files at once. This is a good opportunity to utilize the 'wildcard' `*` syntax. Using a star in place of a filename, or part of a filename, will tell the terminal that you want to run all samples that match the remaining descriptors. For example, `*.fasta` will refer to all files that have the extention .fasta. Additionally, SampleNumber*.fasta will refer to all files that start with 'SampleNumber' and end with the .fasta extension. Using a wildcard will be very helpful when you have a large dataset.
To identify potential arguments for `fastqc`, you can use the `-h` argument:
```
(base) bot3404$ fastqc -h
fastqc -h
FastQC - A high throughput sequence QC analysis tool
SYNOPSIS
fastqc seqfile1 seqfile2 .. seqfileN
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam]
[-c contaminant file] seqfile1 .. seqfileN
DESCRIPTION
FastQC reads a set of sequence files and produces from each one a quality
control report consisting of a number of different modules, each one of
which will help to identify a different potential type of problem in your
data.
If no files to process are specified on the command line then the program
will start as an interactive graphical application. If files are provided
on the command line then the program will run with no user interaction
required. In this mode it is suitable for inclusion into a standardised
analysis pipeline.
The options for the program as as follows:
-h --help Print this help file and exit
-v --version Print the version of the program and exit
-o --outdir Create all output files in the specified output directory.
Please note that this directory must exist as the program
will not create it. If this option is not set then the
output file for each sequence file is created in the same
directory as the sequence file which was processed.
--casava Files come from raw casava output. Files in the same sample
group (differing only by the group number) will be analysed
as a set rather than individually. Sequences with the filter
flag set in the header will be excluded from the analysis.
Files must have the same names given to them by casava
(including being gzipped and ending with .gz) otherwise they
won't be grouped together correctly.
--nano Files come from nanopore sequences and are in fast5 format. In
this mode you can pass in directories to process and the program
will take in all fast5 files within those directories and produce
a single output file from the sequences found in all files.
--nofilter If running with --casava then don't remove read flagged by
casava as poor quality when performing the QC analysis.
--extract If set then the zipped output file will be uncompressed in
the same directory after it has been created. By default
this option will be set if fastqc is run in non-interactive
mode.
-j --java Provides the full path to the java binary you want to use to
launch fastqc. If not supplied then java is assumed to be in
your path.
--noextract Do not uncompress the output file after creating it. You
should set this option if you do not wish to uncompress
the output when running in non-interactive mode.
--nogroup Disable grouping of bases for reads >50bp. All reports will
show data for every base in the read. WARNING: Using this
option will cause fastqc to crash and burn if you use it on
really long reads, and your plots may end up a ridiculous size.
You have been warned!
--min_length Sets an artificial lower limit on the length of the sequence
to be shown in the report. As long as you set this to a value
greater or equal to your longest read length then this will be
the sequence length used to create your read groups. This can
be useful for making directly comaparable statistics from
datasets with somewhat variable read lengths.
-f --format Bypasses the normal sequence file format detection and
forces the program to use the specified format. Valid
formats are bam,sam,bam_mapped,sam_mapped and fastq
-t --threads Specifies the number of files which can be processed
simultaneously. Each thread will be allocated 250MB of
memory so you shouldn't run more threads than your
available memory will cope with, and not more than
6 threads on a 32 bit machine
-c Specifies a non-default file which contains the list of
--contaminants contaminants to screen overrepresented sequences against.
The file must contain sets of named contaminants in the
form name[tab]sequence. Lines prefixed with a hash will
be ignored.
-a Specifies a non-default file which contains the list of
--adapters adapter sequences which will be explicity searched against
the library. The file must contain sets of named adapters
in the form name[tab]sequence. Lines prefixed with a hash
will be ignored.
-l Specifies a non-default file which contains a set of criteria
--limits which will be used to determine the warn/error limits for the
various modules. This file can also be used to selectively
remove some modules from the output all together. The format
needs to mirror the default limits.txt file found in the
Configuration folder.
-k --kmers Specifies the length of Kmer to look for in the Kmer content
module. Specified Kmer length must be between 2 and 10. Default
length is 7 if not specified.
-q --quiet Supress all progress messages on stdout and only report errors.
-d --dir Selects a directory to be used for temporary files written when
generating report images. Defaults to system temp directory if
not specified.
BUGS
Any bugs in fastqc should be reported either to simon.andrews@babraham.ac.uk
or in www.bioinformatics.babraham.ac.uk/bugzilla/
```
Below are some sample command line arguments:
> `fastqc [-o output dir] [-f fastq|bam|sam] seqfile1 .. seqfileN`
### `fastqc activity`
*Try writing a line of code below that prompts FastQC to create an output file.*
```
```
Use CyberDuck to download the FastQC output file. To connect to the SPHAGNUM computer in CyberDuck you will need to open an `sftp` connection, and specify the server as `sphagnum.ttu.edu` with `bot3404` as the username:

and view it in HTML.
**Questions**
1. How many reads are in the files?
2. How many total base-pairs (DNA letters) are in the files?
3. What is the distribution of read lengths?
4. Are there any warnings in FASTQC?
## Flags and Helper Documents
If you find yourself stuck or confused while writing a line of code while using a program, try using the helper printout. For example, FASTQC has a helper document that explains arguments needed for running the program. Access it using `fastqc -h`.
Some programs may use `man program` or `program --help` for their help documents. It is also worth looking for the program's webpage to view recent documentation -- the developer may also have set up a help forum or e-mail list to get assistance.
Other good sources of information for UNIX systems is [StackOverflow](https://stackoverflow.com/). More specific help for bioinformatics can be found using [biostars](https://www.biostars.org/), [seqanswers](http://seqanswers.com/), or the [bioinformatics reddit forum](https://www.reddit.com/r/bioinformatics/).
When in doubt, it is frequently useful to use a Google search with the name of the program and the error message. Often, these searches will take you directly to a post on one of the sites mentioned above!
## Trimming Data
We will use [FastP](https://github.com/OpenGene/fastp) to trim down reads before pairing them. We trim reads for many reasons, such as removing the adapter sequences from a read (adapter sequences are critical to the library prep stage, but cause problems in bioinformatics), or to trim out the low quality, short, sections of reads. We want to keep only high quality and longer reads to input into the pipeline.
> fastp supports both single-end (SE) and paired-end (PE) input/output.
>* for SE data, you only have to specify read1 input by -i or --in1, and specify read1 output by -o or --out1.
>* for PE data, you should also specify read2 input by -I or --in2, and specify read2 output by -O or --out2.
>* if you don't specify the output file names, no output files will be written, but the QC will still be done for both data before and after filtering.
>* the output will be gzip-compressed if its file name ends with .gz
`fastp -i [untrimmed.forward].fq -I [untrimmed.reverse].fq -o [trimmedoutput.forward].fq -O [trimmedoutput.reverse].fq`
SOURCE: [OpenGene/ FastP Github Page](https://github.com/OpenGene/fastp)
In addition to the simple usage of FastP, we also need to add additional arguments for the type of data we have. The instructors will inform you of additional arguments needed based on the data set you use.
### `fastp` activity
*Try adapting the simple usage code for your dataset.*
```
```
## Comparing trimmed and untrimmed output
Previously, we ran FastQC on the untrimmed data.
### Final Activity
*Write a command to run FastQC on the trimmed reads*
```
```
What are the differences between the two outputs?
 Sign in with Wallet
Connect another wallet
Sign in with Wallet
Connect another wallet