---
tags: ggg, ggg2020, ggg298
---
# GGG 298, Jan 2020 - Week 4 - snakemake for running workflows!
[toc]
## Side note
I'm available for office hours today, in CCAH 251 or 253.
Shannon Office Hours: Friday 11am-12pm in the DataLab (3rd Floor Library)
## Learning objectives
By the end of this lecture, students will:
* know how to make basic workflows in snakemake rules
* understand variable substitution in snakemake rules
* understand wildcard matching in snakemake rules
* be introduced to reasons why workflow systems can help you do your computing more easily
## Introduction to workflows
Many things in bioinformatics are workflows. Data goes in, data comes out! Today we're going to talk about ways of automating this, using snakemake.
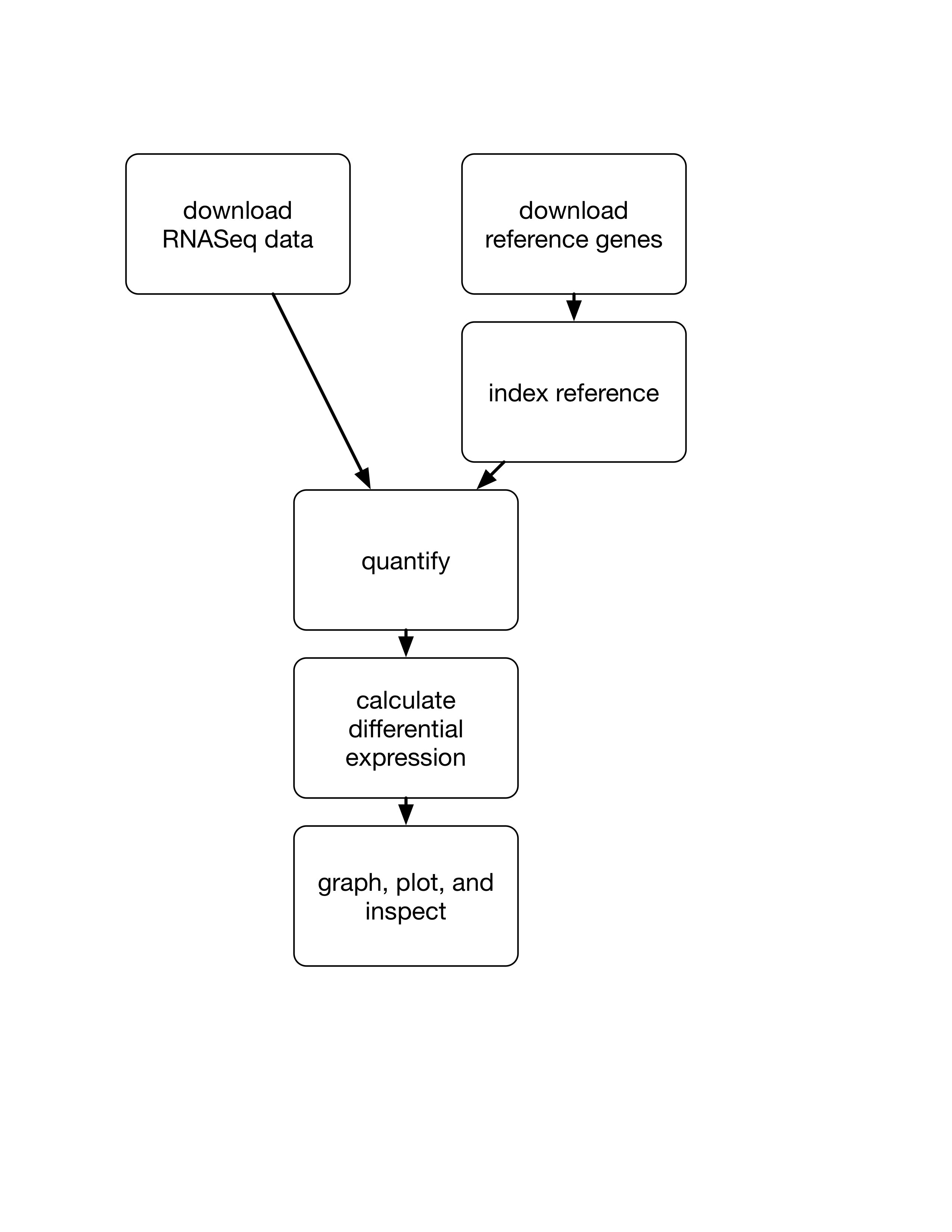
Generally the end result of a workflow is something you load into R and process (e.g. RMarkdown).
Realistically workflows get [much more complicated](https://github.com/spacegraphcats/2018-paper-spacegraphcats/raw/master/paper/figures/hu_dag.pdf) when you're doing real things, too!
### What is a workflow system, and why use one?
Every computational workflow consists of multiple steps, taking previous outputs and/or data/information in and executing upon them and outputting something.
raw data -> workflow -> summary data for plotting and statistics
See also [Nature Toolbox, "Workflow systems turn raw data into scientific knowledge"](https://www.nature.com/articles/d41586-019-02619-z)
## Introduction to snakemake
[snakemake](https://snakemake.readthedocs.io/) is an awesome workflow system created by Johannes Koester and others (see [2012 publication](https://academic.oup.com/bioinformatics/article/28/19/2520/290322)). Other workflow systems work similarly but have different syntax; while we use snakemake, we can also recommend [nextflow](https://www.nextflow.io/), [Common Workflow Language](https://www.commonwl.org/), and [Workflow Definition Language](https://github.com/openwdl/wdl). The latter two are more for workflow engineers than the first two.
snakemake works by looking at a file, called a Snakefile, that contains recipes for creating files. Then it uses the recipes and command line options to figure out what needs to be created and how to do it, and then ...runs it!
So, it's (mostly) in the Snakefile, folks...
The name 'snakemake' comes from the fact that it's written in (and can be extended by) the Python programming language. (Confusingly, Python is actually named after Monty Python, not the reptiles, but whatever.)
### What we're going to do today
* start with a snakemake workflow for calculating fastqc analyses of multiple FASTQ data files
* add a snakemake workflow for RNAseq quantification of samples
* iterate on this workflow a few times!
### Setting ourselves up
Go ahead and log into the farm.
Create a working directory:
```
mkdir -p ~/298class4
cd ~/298class4
```
Download some data:
```
curl -L https://osf.io/5daup/download -o ERR458493.fastq.gz
curl -L https://osf.io/8rvh5/download -o ERR458494.fastq.gz
curl -L https://osf.io/xju4a/download -o ERR458500.fastq.gz
curl -L https://osf.io/nmqe6/download -o ERR458501.fastq.gz
```
Configure bioconda channels, just in case this didn't happen the first time through:
```
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
```
(rerunning these commands is harmless!)
and, finally, activate our previous conda environment that contains fastqc. Then install salmon:
```
conda activate fqc
conda install -y -n fqc salmon
```
(if you don't have a fqc environment from [week 3](https://hackmd.io/7XxZCUQkRiOq0QnOHDZtaQ?view), you can create this with `conda create -y -n fqc -c bioconda fastqc salmon`.)
We are now set!
### A basic snakemake recipe: FASTQC reports
We have a file "ERR458493.fastq.gz" and we want to make "ERR458493_fastqc.html" and "ERR458493_fastqc.zip" from it.
We _could_ run `fastqc ERR458493.fastq.gz` at the command line, but let's use this task as the start of our snakemake workflow first!
Run:
```
nano -ET4 Snakefile
```
and copy/paste the following in;
```
rule all:
input:
"ERR458493_fastqc.html",
"ERR458493_fastqc.zip"
rule make_fastqc:
input:
"ERR458493.fastq.gz"
output:
"ERR458493_fastqc.html",
"ERR458493_fastqc.zip"
shell:
"fastqc ERR458493.fastq.gz"
```
hit <kbd>CTRL+X</kbd>, <kbd>y</kbd>, <kbd>ENTER</kbd> to save the changes.
now run:
```
snakemake -p
```
and you will see snakemake run fastqc for you, as specified!
The logic above is this:
* each **rule** tells Snakemake how to do _something_
* the first rule (in this case called `all`) is the rule run by default, so we are asking snakemake to create the two target files `ERR458493_fastqc.html` and `ERR458493_fastqc.zip`.
* snakemake first looks at the directory to see if the target files are there. (They're not!)
* snakemake then looks at the rest of the rules (in this case, there's only one!) to see if it can figure out how to make the target files.
* the `make_fastqc` rule says, "if this input exists, you can run the provided shell command to make that output".
* so snakemake complies!
Here, the "input:" in the rule `all` _has_ to match the "output" in the rule `make_fastqc` or else Snakefile wouldn't know what to make.
**Meta-notes:**
* Snakefile contains a snakemake workflow definition
* the rules specify steps in the workflow
* You can "decorate" the rules to tell snakemake how they depend on each other.
* rules can be in any order, but put "default goals" as first
* information within rules such as input and output (and other things) can be in any order, as long as they are before shell.
* these are just shell commands, with a bit of "decoration". You could run them yourself if you wanted!
* rule names are arbitrary (letters, numbers, _)
* you can specify a subset of outputs, e.g. just the .html file, and snakemake will run the rule even if it only needs one of the files.
* it goes all red if it fails! (try breaking one command :)
* it's all case sensitive
* tabs and spacing matters... that's why we're using `nano -ET4` to edit!
* you can make lists for multiple input or output files by separating filenames with a comma
### Some features of workflows
If you run `snakemake -p` again, it won't do anything. That's because all of the input files for the first rule already exist!
However, if you remove a file and run snakemake:
```
rm ERR458493_fastqc.html
snakemake -p
```
then snakemake will run fastqc again, because now you don't have one of the files you're asking it to make!
This ability to selectively figure out whether or not to run a command is one of the most convenient features of snakemake.
### Add a variable substitution
Let's make the `make_fastqc` rule a little more generic. Use `nano -ET4 Snakefile` to edit the file and make the rule look like this:
```
rule make_fastqc:
input:
"ERR458493.fastq.gz"
output:
"ERR458493_fastqc.html",
"ERR458493_fastqc.zip"
shell:
"fastqc {input}"
```
What does this do? It replaces the `{input}` with whatever is in the "input:" line, above.
### EXERCISE: add a new rule
TODO: Add a new rule, called `make_fastqc2`, that runs fastqc on ERR458501.fastq.gz
Does it run?
Reminder: remember to add the desired output file to the "all" rule as an input, too!
### Add wildcards
You should now have two rules, `make_fastqc` and `make_fastqc2`, that have the _same_ shell: command. But they have different inputs and outputs: one has "ERR458493.fastq.gz" as an input, and "ERR458493_fastqc.html" and "ERR458493_fastqc.zip" as outputs, while the other has "ERR458501.fastq.gz" as an input, and "ERR458501_fastqc.html" and "ERR458501_fastqc.zip" as outputs. If you line these up, you'll notice something interesting:
```
ERR458493.fastq.gz
ERR458493_fastqc.html
ERR458493_fastqc.zip
^^^^^^^^^
ERR458501.fastq.gz
ERR458501_fastqc.html
ERR458501_fastqc.zip
^^^^^^^^^
```
Notice how the top three and the bottom three have the same prefix (ERR458), and how the suffixes are the same between the two samples?
Use `nano -ET4 Snakefile` and change the `make_fastqc` rule so:
* the input: is `"{sample}.fastq.gz"`
* the output is `"{sample}_fastqc.html", "{sample}_fastqc.zip"`
* delete the `make_fastqc2` rule (you can use CTRL-K to delete an entire line if your cursor is on that line).
Your complete Snakefile should look like this:
```
rule all:
input:
"ERR458493_fastqc.html",
"ERR458493_fastqc.zip",
"ERR458501_fastqc.html",
"ERR458501_fastqc.zip"
rule make_fastqc:
input:
"{sample}.fastq.gz"
output:
"{sample}_fastqc.html",
"{sample}_fastqc.zip"
shell:
"fastqc {input}"
```
This tells snakemake that _any time_ it is asked for a file that ends with `_fastqc.html` or `_fastqc.zip`, it should look for a similarly named file that ends with `.fastq.gz` and, if it finds one, it can run `fastqc` on that file to produce those outputs.
Try it!
```
rm *.html
snakemake -p
```
Note that wildcards print out in the snakemake output for each task, so you can see exactly what is being substituted!
Final note: wildcards operate entirely within a single rule, not across rules.
### EXERCISE: Add more fastqcs
Update the Snakefile so that it runs fastqc on "ERR458494.fastq.gz" and "ERR458500.fastq.gz" too.
### Reprise and summary: what we did
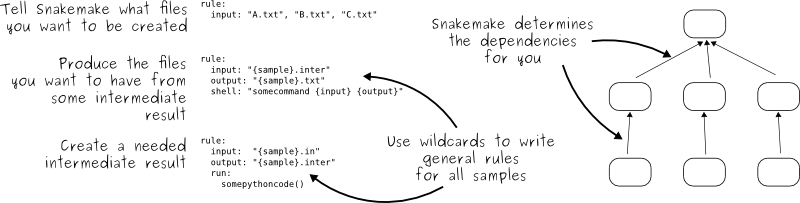
### EXERCISE: Add some more rules
Now let's add some more rules, at the bottom, so that we can do the quantification.
We need to do three things: (1) download a reference transcriptome, (2) index the reference transcriptome, and (3) quantify the reference genes based on the reads (using salmon).
Let's add the first two as new rules. Here are the shell commands to run; can you make two new rules from them? one called `download_reference` and the other called `index_reference`?
the `download_reference` shell command is:
```
curl -O https://downloads.yeastgenome.org/sequence/S288C_reference/orf_dna/orf_coding.fasta.gz
```
the `index_reference` shell command is:
```
salmon index --index yeast_orfs --transcripts orf_coding.fasta.gz
```
(note that you can always just run these commands at the prompt if you want to see if they work!)
Suggestion:
* start by creating the rules with only a `shell:` line. To do this, copy the format of a previous rule! Note that you can put in empty input: and output: lines if you like.
* then, add an `output:` line to `download_reference`. What file does it produce?
* (note that `download_reference` doesn't take any input files, because it's downloading the data from the Internet!)
* then, add an `input:` line to `index_reference`. What does it take in?
* in `index_reference`, replace the filename it takes in the shell line with `{input}`, too.
* now, run the commands via snakemake to see if they work -- `snakemake -p `
**(one) Solution**
Add these to the end of your Snakefile
```
rule download_reference:
output:
"orf_coding.fasta.gz"
shell:
"curl -O https://downloads.yeastgenome.org/sequence/S288C_reference/orf_dna/orf_coding.fasta.gz"
rule index_reference:
input:
"orf_coding.fasta.gz"
output:
"yeast_orfs"
shell:
"salmon index --index {output} --transcripts {input}"
```
BUT if you try to run `snakemake -p` then it won't run... we have to specify the rule to run:
```
snakemake -p download_reference
snakemake -p index_reference
```
If you'd like to have `snakemake -p` run these, then add their output to the input: of `rule all:`
Alternatively, you can ask for the filenames: `snakemake -p yeast_orfs orf_coding.fasta.gz`.
## Running salmon quant & working with multiple samples
The next command we want to snakemake-ify is this one:
```
for i in *.fastq.gz
do
salmon quant -i yeast_orfs --libType U -r $i -o $i.quant --seqBias --gcBias
done
```
we _could_ do this by copying the command above into a "shell:"" block, like so:
```
rule salmon_quant:
...
shell:
"""for i in *.fastq.gz
do
salmon quant -i yeast_orfs --libType U -r $i -o $i.quant --seqBias --gcBias
done"""
```
_but_ this is not ideal, for a few reasons.
The two main reasons are these:
1. it doesn't permit fine-grained error reporting -- if one of the `salmon quant` commands fails, the entire rule fails, so all four need to be re-run.
2. the time to run the for loop will scale with the number of samples to run: snakemake can't help you by running the commands in parallel.
a third reason is that, as written, the command cannot easily take advantage of snakemake wildcards (like `{sample}` or `{input}`).
(These are all "bad smells", i.e. if you find your Snakefile is behaving like this, it's time to reconsider your approach!)
These are all related to the feature that snakemake specifies workflows _declaratively_, as recipes, and can then choose how to run them based on the resources its given.
**Analogy time:** if you have a kitchen, and a chef, you can either give them a recipe procedure and ask them to follow it; or you can give them a recipe book, and then tell them what dishes you want for dinner. In the former case, they are stuck doing things in the order you specify. In the latter case, they have the freedom to do things in the order that is convenient or efficient. And, if you hire more chefs, you may be able to produce meals faster and more efficiently!
(We'll discuss *how* to provide more chefs to snakemake below, in more detail.)
## So, how do we do multiple samples?!
With more wildcards, like we did in the fastqc example, above!
First, let's start with a test case by writing a single sample rule:
```
rule salmon_quant:
shell:
"salmon quant -i yeast_orfs --libType U -r ERR458493.fastq.gz -o ERR458493.fastq.gz.quant --seqBias --gcBias"
```
and make sure that works: `snakemake -p salmon_quant`
Now, add `input:` and `output:`...
```
rule salmon_quant:
input: "ERR458493.fastq.gz"
output: "ERR458493.fastq.gz.quant"
shell:
"salmon quant -i yeast_orfs --libType U -r ERR458493.fastq.gz -o ERR458493.fastq.gz.quant --seqBias --gcBias"
```
and check that for syntax: `snakemake -p salmon_quant`
and finally replace the prefixes with the `{sample}` `{input}` and `{output}` wildcards we learned before:
```
rule salmon_quant:
input: "{sample}"
output: "{sample}.quant"
shell:
"salmon quant -i yeast_orfs --libType U -r {input} -o {output} --seqBias --gcBias"
```
Now, you can no longer run `salmon_quant` directly -- you have to ask snakemake for a particular file. Try:
```
rm -fr ERR458493.fastq.gz.quant
snakemake -p ERR458493.fastq.gz.quant
```
The reason for this is that snakemake doesn't *automatically* look at all the files in the directory and figure out which ones it can apply rules to - you have to ask it more specifically, by asking for the specific files you want.
Last but not least, we need to add this info into the default rule.
### EXERCISE: make the command `snakemake` run with no arguments for all four `salmon quant` commands.
### Another random aside: `-n`
If you give snakemake a `-n` parameter, it will tell you what it thinks it should run but won't actually run it. This is useful for situations where you don't know what needs to be run and want to find out without actually running it.
### Concrete vs abstract rules, and debugging advice
Rules that only mention specific files (no wildcards!) are *concrete* rules. You have to provide snakemake with at least one specific file to make, or it will tell you that there's a `WorkflowError`:
>WorkflowError:
Target rules may not contain wildcards. Please specify concrete files or a rule without wildcards.
You can provide a filename on the command line, or you can put one in a rule and run that rule, but you have to provide at least one concrete request.
In contrast, rules containing wildcards are _abstract_ rules, and snakemake will use them as needed to build the concrete files requested.
To debug WorkflowErrors as above,
1. try running your first rule (in the workflow, not in the Snakefile) by asking snakemake to build a file that that rule should produce.
2. iterate until you find a rule that doesn't produce the requested file, and then fix that rule.
3. if you can't find such a rule, then your workflow works :)
## Intermediate snakemake: what comes next?
There are many advanced features to snakemake, and we'll touch on a few of them here.
### Rule-specific conda environments with `conda:` and `--use-conda`
If you specify a [conda environment file](https://hackmd.io/7XxZCUQkRiOq0QnOHDZtaQ?view#Making-and-using-environment-files), in an `conda:` block in a rule, and run conda with `--use-conda`, it will always run that rule in that software environment.
This is useful when you want to version-pin a specific action, and/or have conflicting software in different rules.
### parallelizing snakemake: -j
You can tell snakemake to run things in parallel by doing
```
snakemake -j 2
```
This will tell snakemake that it can run up to two jobs at a time. (It automatically figures out which jobs can be run at the same time by looking at the workflow graph.)
We'll show you how to do this on a larger scale on a cluster in a few weeks :)
### More things we will discuss ...later
* glob and building off of files in the current directory
* glob, expand, and parallelism
* params
* config files
## Advanced snakemake
R integration
Python integration
## Practical advice: How to build your workflow
General advice:
* start small, grow your snakefile!
* DO copy and paste from this tutorial and others you find online!
* it rarely hurts to just re-run snakemake until it does nothing but error out, and then analyze that error :)
* we can help, at [Meet and Analyze Data! Wed 3-5pm](http://mad.oxli.org), or [online](http://mad.oxli.org).
### Approach 1: write down your shell commands
Pick a small, linear workflow, and then:
* make rules for each of your shell commands, and run them individually;
* add input and output to each rule until you can "just" run the last rule and have it all work;
* start adding wildcards as you see fit!
### Approach 2: automate one step that you run a lot
alternatively, if you have a complex workflow that would take a lot of time and energy to convert,
* pick a specific part that you would like to run on a lot of files!
## Why use a workflow manager?
what do (snakemake) workflows do for me?
A laundry list:
- declarative vs procedural specification
- allows analysis of workflow graph, parallelization
- allows declaration of specific software needed, for later tracking
- can compare workflows more easily, in theory (=> reproducibility)
- can rerun failed steps
- tracks dependencies on files
- allows rerunning just the bits that need to rerun
- reusability, in theory
- different conda environments for each step
- allows pinning of software versions
- allows use of potentially incompatible software
For me, the main reason to use snakemake is that it lets be sure that my workflow completed properly. snakemake tracks which commands fails, and will stop the workflow in its tracks! This is not something that you usually do in shell scripts.
(It turns out that I've spent a lot of my life being a bit paranoid about whether my commands actually ran correctly!)
## Dealing with complexity
Workflows can get really complicated; [here](https://github.com/spacegraphcats/2018-paper-spacegraphcats/blob/master/pipeline-base/Snakefile), for example, is one for our most recent paper. But it's all just using the building blocks that I showed you above!
If you want to see some good examples of how to build nice, clean, simple looking workflows, check out [this RNAseq example](https://github.com/snakemake-workflows/rna-seq-star-deseq2).
## Summary of what we did today.
* Snakefiles contain rules
* snakemake uses those rules to figure out what files to build
* the basic idea is simple, but there are lots of tricks that we will teach you!
## More Snakemake resources
google is your friend!
The first three 201(b) class materials are a fairly gentle introduction: https://hackmd.io/YaM6z84wQeK619cSeLJ2tg#Schedule-of-lab-topics
here's another tutorial I wrote... [link](https://github.com/ctb/2019-snakemake-ucdavis/blob/master/tutorial.md)
a free book! -- [the Snakemake book](https://endrebak.gitbooks.io/the-snakemake-book/chapters/hello_world/hello_world.html)
angus 2019 material -- [Workflow Management using Snakemake](https://angus.readthedocs.io/en/2019/snakemake_for_automation.html)
### Dealing with complexity
Workflows can get really complicated; [here](https://github.com/spacegraphcats/2018-paper-spacegraphcats/blob/master/pipeline-base/Snakefile), for example, is one for our most recent paper. But it's all just using the building blocks that I showed you above!
If you want to see some good examples of how to build nice, clean, simple looking workflows, check out [this RNAseq example](https://github.com/snakemake-workflows/rna-seq-star-deseq2).
## Debugging Q: What if I get a workflow error?
If you get the error
>WorkflowError:
>Target rules may not contain wildcards. Please specify concrete files or a rule without wildcards.
>
then what's happening is you're trying to directly run a rule with a wildcard in it (i.e. an abstract rule), and snakemake can't figure out what the wildcard should be; instead, ask snakemake to build a specific file.
For example, with the rule immediately above that adds wildcards to `map_reads`,
```
snakemake map_reads
```
will complain about the target rule containing wildcards. You should instead run
```
snakemake SRR2584857_1.sam
```
which snakemake can use to figure out what the wildcards should be.
An alternative to specifying the file on the command line is to put it in the default rule, e.g. `rule all:` (see [the section on default rules](https://hackmd.io/cGYzxz07SseGxH0y2gjYJw?view#Create-a-good-%E2%80%9Cdefault%E2%80%9D-rule)) and then you can run `snakemake`.
We can use the `-n` gives a dry run of the Snakefile. For example `snakemake -p -n`
### A quick checklist:
* Are you asking snakemake to create a _specific_ file?
* either by executing `snakemake <filename>`
* or by specifying a rule that has an input or an output without wildcards,
* or by providing a default rule?
* for any rule that you expect to be executed automatically (because some other rule needs its output), have you specified `output:`?