# Analysis of gene function during butterfly wing development
BI332 Developmental Biology - Fall 2019
## Background
Experimental manipulations are an important tool to investigate how patterns are created during development. By disrupting specific elements of a developing system, we can infer the normal function of that element from changes in the phenotype. Lose-of-function experiments of this kind can be done by removing cells from an embryo or juvenile, but also by removing the function of genes.
Historically, genetic control of development was studied by mutagenesis. In this method, mutations are created at random throughout the genome by chemical mutagens or X-ray damage. Many individuals need to be screened for a phenotype altering the structure or developmental process under study. Then the affected gene must be mapped and characterized at the level of its DNA sequence. That process is laborious and prone to bias at levels of mutation and screening.
### CRISPR/Cas9
CRISPR/Cas9 gene editing is a method that can be used to permanently alter DNA in live cells at specific targeted sequences. It relies on Cas9, which evolved for viral defense in the bacterium *Streptococcus pyogenes*. Cas9 is an endonuclease, which can cut the phosphodiester bonds on both strands of DNA, producing a double-strand break. Unlike other endonucleases, Cas9 is directed to cut DNA by a short guide RNA. In *Strep. pyogenes* this guide consists of two separate RNAs. A tracrRNA has a sequence that is recognized and bound by Cas9. This component is generic. However, a crRNA has a 3$^\prime$ region of base complementary to the tracrRNA and a 5$^\prime$ sequence that targets the Cas9/RNA complex to a specific DNA sequence. In *Strep. pyogenes* the crRNA sequence is derived from viral genes that have previously infected the cell. In this way the bacterium is able to recognize and destroy new, invading viral DNA ([Barrangou 2015](https://www.ncbi.nlm.nih.gov/pubmed/25574773)).

> **Figure 1.** In *Streptococcus pyogenes* Cas9 uses a generic tracrRNA and a target-specific crRNA to identify and destroy viral DNA. (Image source: https://sg.idtdna.com/jp/)
The *Strep. pyogenes* immune mechanism has been modified to become a versatile tool of molecular genetics. *Cas9* has been cloned into *E. coli* were the protein can be grown and produced in large quantities. To increase its effectiveness in eukaryotic cells, most researchers use a modified version of the protein that includes a eukaryotic nuclear-localization signal (NLS) peptide at the N-terminus of the protein. While it is possible to use a crRNA and tracrRNA, many researchers use a single short guide RNA (sgRNA) to simplify the process. The guide RNA must retain a specific 80-nucleotide sequence at its 3$^\prime$ end, which is bound by Cas9: 5$^\prime$-GUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUU-3$^\prime$. At the 5$^\prime$-end almost any 20-nucleotide sequence can be used to target Cas9 to a position in the genome. There Cas9 will cut leaving a 1-bp overhang, 17-nucleotides from the 5$^\prime$-end of the guide's sequence ([Zou & Liu 2016](https://www.nature.com/articles/srep37584)). Most animal cells will repair double-strand breaks of this kind using a mechanism called non-homologous end joining (NHEJ). However doing so produces a small deletion, at least 1 bp but sometimes 100's of bp. If the target is within the coding sequence of a gene, these mutations may cause a frame-shift that renders the gene non-functional.

> **Figure 2.** Most applications of CRISPR/Cas9 for gene editing now use a single guide RNA (usually abbreviated sgRNA, but labeled gRNA in this figure). This guide targets Cas9 to a specific target DNA sequence, where it makes a double-strand break, which can then be repaired by NHEJ, albeit with a high frequency of intel mutations. (Image source: https://www.addgene.org/crispr/guide/)
#### Short guide RNA design
With the rise of CRISPR/Cas9 gene editing, guide design has become a useful skill and a many companies charge to provide algorithms that automate the process. However, the basic rules are straight forward. The only major constraint on the selection of a target DNA sequence is necessity of a protospacer adjacent motif (PAM). Enzymes similar to Cas9 have evolved in several different bacterial species, where the PAM sequence differs. However in the most commonly used commercial versions of Cas9, derived from *Strep. pyogenes*, the PAM sequence is 5$^\prime$-NGG-3$^\prime$ (where "N" is any base followed by two guanines). This PAM sequence must appear immediately downstream of the targeted sequence in the genome. (However, the PAM sequence does not appear in the guide RNA.)

> **Figure 3.** And example of how Cas9 and an sgRNA are positioned relative to a target DNA and the PAM sequence. (Image source: https://web.science.uu.nl/developmentalbiology/)
People working with CRISPR in diverse organisms have also developed a set of other suggestions for designing sgRNAs (e.g. [Zhang & Reed 2017](https://www.researchgate.net/publication/319369908_A_Practical_Guide_to_CRISPRCas9_Genome_Editing_in_Lepidoptera/fulltext/59a74da3a6fdcc61fcfbd675/A-Practical-Guide-to-CRISPR-Cas9-Genome-Editing-in-Lepidoptera.pdf); [Addgene 2017](https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing); Martin & O'Connell in press).
- Avoid target sites close to start codon of a gene. In some cells, some genes may be able to use alternative ATG start codons downstream of the annotated start codon.
- Similarly, avoid target sites close to the stop codon of a gene to maximize the chances of creating a non-functional allele.
- If you are working from transcript sequence, there is a risk that the designed target sequence will span an exon-exon junction. If so, the guide RNA is unlikely to bind the genome.
- A guide sequence can be 17-27 nucleotides, on either strand. But 20 nt may be optimal.
- Watch out for N's in your sequence. Do not choose sequences that include N's.
- Never use a guide with 3 or more U’s in a row. This sequence can act as an RNA polymerase III terminator.
- The GC content of the target sequence should be between ~ 30-80%. Higher GC content, especially at the 3$^\prime$ end may increase editing efficiency, since it increases the binding strength of the sgRNA with the DNA. However, targets that are very high in %GC may be unlikely to open in the first place!
- Avoid additional G‘s after the PAM.
- If sgRNAs will be transcribed from a PCR product of plasmid, be sure there is a 5$^\prime$-GG sequence. If one does not naturally occur in the target sequence, just add it anyway. This sequence increases the efficiency of transcription from a T7 promoter. Any mismatch at the 5$^\prime$-end will not negatively impact the guide's binding to the target DNA.
### The butterfly wing as a model for tissue patterning
In developmental biology, patterning is the process by which initially equivalent cells adopt different fates. While there are many contexts in which this takes place, some systems have been studied extensively as generalizable models. The appendages or insects and vertebrates are classical systems for the study of patterning. Studies of pigmentation patterning in diverse animals have recently become increasingly common.

> **Figure 4.** The painted lady butterfly, *Vanessa cardui*, showing the ventral side of its wings. (Image source: WikiMedia)
In this course, we will use the wings of the painted lady butterfly, *Vanessa cardui*, as a model to study patterning. This system has many advantages. Caterpillars are commercially available throughout the year, and the entire life cycle can be completed in captivity. Newly laid eggs hatch and develop to adulthood in 5 weeks. This species is also native to New England, so (while we will not plan to release any animals) there is limited ecological risk from accidental release. Importantly, a [transcriptome is available](http://www.butterflygenome.org/?q=node/4) for this species and a [web portal has been constructed for BLAST search](http://www.butterflygenome.org/?q=node/5) of those sequences. The development of butterfly wings has also been studied in some detail, provided some basic context.

> **Figure 5.** The generalized anatomy of nymphalid butterfly wing patterns. Pattern elements are organized in parallel. From distal to proximal, there are marginal and submarginal bands (chevrons in some species), the border ocelli (sometimes enlarged to form eyespots), and the central and basal bands. These pattern elements are repeated in the individual wing cells, which are bordered by veins. Pattern elements are thought to share developmental mechanisms, but are known to evolve separately. (Image source: [Beldade & Brakefield 2002](https://www.ncbi.nlm.nih.gov/pubmed/12042771))

> **Figure 6.** The wings of *Vanessa cardui*. Dorsal surfaces are in the top row; ventral surfaces are the bottom row. Males are on the left; Females on the right. Females tend to be larger than males, but there is no reliable pattern to distinguish the sexes. (Image source: http://www.raisingbutterflies.org/painted-lady/)
#### Genes with known roles in butterfly wing patterning
- **WntA** encodes a morphogen that organizes much of the pattern across the wing ([Mazo-Vargas et al., 2017](https://www.pnas.org/content/114/40/10701)). CRISPR experiments in *V. cardui* targetting *WntA* have bee quite successful, including in undergraduate courses (Martin & O'Connell, in press).
- ***Distal-less* (*Dll*)** encodes a transcription factor required for the development of distal appendages in most animals. It is also known to be required for the border ocelli and marginal elements of the butterfly wing. *Dll* loss-of-function produces different phenotypes depending on the part of the protein that is targeted ([Zhang & Reed, 2016](https://www.nature.com/articles/ncomms11769); [Connahs et al., 2019](https://dev.biologists.org/content/146/9/dev169367)).
- ***optix*** encodes a protein required for orange pigmentation ([Zhang et al., 2017a](https://www.pnas.org/content/114/40/10707)).
- Several genes with roles in melanic pigment production are known or strongly predicted to have role in wing pigmentation, including the transcription factor ***spineless***, and the enzymes encoded by *Dopadecarboxylase* (***Ddc***), ***yellow***, ***ebony***, and ***black*** ([Zhang & Reed, 2016](https://www.nature.com/articles/ncomms11769); [Zhang et al., 2017b](https://www.genetics.org/content/205/4/1537)).
#### Additional candidate genes
- **EGF** signaling is important in the development of insects wings, especially in veins. Since the placement of veins is key the patterning of butterfly wings, it is possible EGF signaling is involved in this process.
- The **atypical cadherin pathway**, also known by the names of two of its components **fat** and **Hippo**, controls planar cell polarity (the orientation of epithelial cells) in the wings of *Drosophila*. Since each scale in the wing of a butterfly is homologous to the bristles of flies, it is possible that atypical cadherin signaling plays a role in patterning butterfly wings.
- ***doublesex*** (***dsx***) encodes a transcription factor that is a key regulator of somatic sex determination in insects.
## Procedures
### Design of sgRNAs
#### For lab on September 26.
1. With you lab partner and instructor, **choose a gene to target** for CRISPR. Ideally, lab groups will chose different genes, with some groups choosing "safe" genes, with a history of successful mosaic gene knockout, and others choosing unproven targets.
2. **Obtain the sequence of your target gene**.
- Open a web browers to https://www.ncbi.nlm.nih.gov/protein/
- Enter the name of your gene and the word "Lepidoptera".
- From the list of responses, choose one that is relatively long, and if possible from a common model organism, such as [*Bombyx mori* (the silkmoth)](https://en.wikipedia.org/wiki/Bombyx_mori), *[Manduca sexta* (the tobacco hornworm moth)](https://en.wikipedia.org/wiki/Manduca_sexta, [*Bicyclus anynana*](https://en.wikipedia.org/wiki/Bicyclus_anynana, or [*Eumeta japonica* (the bagworm moth)](https://www.inaturalist.org/taxa/564044-Eumeta-japonica). Record the GenBank accession number and the species in your notes. Then download the amino acid sequence in FASTA format. This can be done by clicking "Send to:". Choose "File" and "FASTA", and click "Create File". This will download the file to your computer.
- Open the file and look at it. FASTA is a simple format for biological sequence data.
- Save and rename the file with a descriptive names, like `Eumeta japonica EGFR protein.fa`
- Next go to http://www.butterflygenome.org/ and open the BLAST tab.
- Copy and paste the amino acid sequence from your orthologous protein into the search window.
- Check the box "V._cardui_unigenes" at the bottom of the window and click "TBLASTN". The results will list *Vanessa cardui* transcripts with sequence similarity to your search query, ranked by their E-value. This metric is analogous to a p-value in statistics, and lower E-values generally indicate a better sequence match.
- Click on the best match (or matches) to see the alignment. Then click on the link to the right that says "FASTA" to download the *Vanessa cardui* transcript nucleotide sequence (as DNA).
- Open the new file and look at it. Save this file with a descriptive name, like `Vanessa cardui c21619_g1 EGFR.fa`
3. **Select an sgRNA for your gene**
- Open Geneious on your computer. Lab computers should already have Geneious installed. If you're working from a personal laptop, you can download a free 14-day trial from https://www.geneious.com/free-trial/
- If Geneious asks for a license key, go to https://www.geneious.com/academic/ to request one.
- Drag and drop your *V. cardui* transcript sequence into the Geneious window.
- From the "Annotate & Predict" menu, select "Find ORFs" adjust the minimum size and apply only the longest and shortest reasonable ORFs (open reading frames). Save the object.
- From the "Tools" menu, select "Cloning" and "Find CRISPR Sites". The default parameters should be ideal, but feel free to discuss them and try other options. Geneious will annotate the sequence with potential sgRNA target sequences. These are color coded, with green being its strongest recommendations. (Be aware that Geneious will include the PAM sequence in the annotation it highlights.)
- Consider these options, in light of the sgRNA design advice above. Discuss the options with your lab partner and instructor. You can mouse-over the annotations to see details like GC content.
- Choose two sgRNA sequences. In your notes, record the guide sequence (5' to 3'), note the position of the end points in your sequence and the strand containing the sequence.
4. Send your sgRNA target sequences to Dr.A.
- In an email to dave.angelini@colby.edu include the following information:
- your target gene's name,
- the name of the species and GenBank accession you used to find the *V. cardui* transcript,
- the cluster and gene ID number of the *V. cardui* transcript (e.g. c21619_g1)
- the sequences of your chosen sgRNA targets, in order of preference,
- the strand on which these guide sequences are found (forward or reverse),
- and the 5' position of each guide sequence.
- Do not include the PAM in your sequences.
- Also attach the ortholog and *V. cardui* transcript FASTA files to the email.
After this week's lab, Dr.A. will place orders for DNA oligos that you will use in the synthesis of sgRNAs.
### Preparation of sgRNAs
It is possible to make sgRNAs in vitro, using a simple, low cost method. After a guide sequence is designed, it can be flanked by a 5$^\prime$ [T7 RNA polymerase](https://en.wikipedia.org/wiki/T7_RNA_polymerase) promoter sequence (taatacgactcactataggg) and 20-nucleotides of the Cas9 recognition sequence (gttttagagctagaaatagc). This oligo can act as one of two primers in a PCR reaction. The second primer has the complementary sequence to the Cas9 backbone, including the 20-nucleotides in the custom oligo. Therefore the two primers have 20-bp of complementarity and overlap. The two primers are used in a PCR reaction (with no other template). The result is a double-stranded DNA with a 5$^\prime$ T7 promoter sequence and the complete Cas9 sgRNA targeting the selected gene-specific sequence. Finally, this DNA served as a template for in vitro RNA synthesis using T7 RNA polymerase.
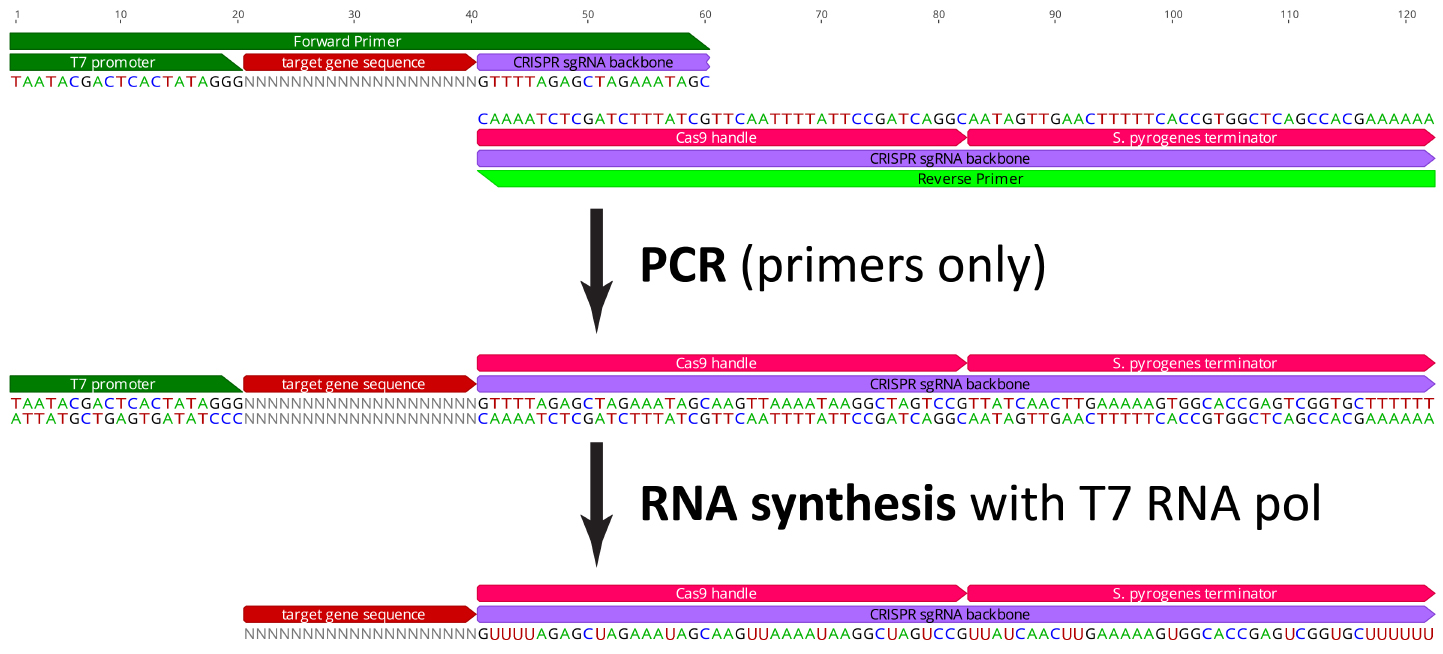
> **Figure 6.** The reagents and steps for synthesis of an sgRNA.
#### primers
| group | gene | *Vc* transcript | position | strand | primer name | primer sequence |
|:--- |:--- |:--- |:--- |:--- |:--- |:--- |
| Emily & Bia | WntA | c24280_g1 | 712 | forward | T7-Vc-WntA-712-Cas9sg | taatacgactcactatagggTCTTTAAGTACAAGATACGAgttttagagctagaaatagc |
| Emily & Bia | WntA | c24280_g1 | 354 | forward | T7-Vc-WntA-354-Cas9sg | taatacgactcactatagggATTCGTCCATGCCCTCTCGGgttttagagctagaaatagc |
| Molly & Catie | Hippo | c26532_g2 | 687 | forward | T7-Vc-Hpo-687-Cas9sg | taatacgactcactatagggCTTGCCTTCCGCCATCTCCAgttttagagctagaaatagc |
| Molly & Catie | Hippo | c26532_g2 | 756 | reverse | T7-Vc-Hpo-756-Cas9sg | taatacgactcactatagggCTTGCCTTCCGCCATCTCCAgttttagagctagaaatagc |
| Yash & Trisha | optix | c27904_g2 | 463 | reverse | T7-Vc-optix-463-Cas9sg | taatacgactcactatagggACAGCGCCTGAAGCTTCGCGgttttagagctagaaatagc |
| Yash & Trisha | optix | c27904_g2 | 590 | forward | T7-Vc-optix-590-Cas9sg | taatacgactcactatagggACTGTTTCAAGGAGCGAACGgttttagagctagaaatagc |
| Jacqueline & Brigid | EGFR | c21619_g1 | 364 | forward | T7-Vc-EGFR-364-Cas9sg | taatacgactcactatagggCCGCAACTACAAATCATTCGgttttagagctagaaatagc |
| Jacqueline & Brigid | EGFR | c21619_g1 | 1041 | forward | T7-Vc-EGFR-1041-Cas9sg | taatacgactcactatagggTTGCCCGAAGACTTGCCGTGgttttagagctagaaatagc |
| Marilyn & Anya | spinless | c25328_g7 | 79 | forward | T7-Vc-ss-79-Cas9sg | taatacgactcactatagggATGTTTCTACAGGCTCTCAAgttttagagctagaaatagc |
| Marilyn & Anya | spinless | c25328_g7 | 196 | reverse | T7-Vc-ss-196-Cas9sg | taatacgactcactatagggGATGGACTATATCTGACTGAgttttagagctagaaatagc |
#### Working with RNA
Remember, single-strand RNA is prone to degradation by RNase, which can be ubiquitous in the environment. As you prepare RNA, take precautions that will preserve your efforts.
- Keep reagents cold. Let tubes thaw on ice, and keep your reactions on ice until they are ready for incubation at higher temperatures.
- Always use filtered pipette tips to prevent introduction of RNase
- Be sure that water used in your reactions is reagent-grade and nuclease-free.
- Wear clean gloves. Change them if you do something that may expose your hands to RNase, like if you touch a computer, phone or notebook.
- Keep tubes containing your reagents and reactions open as breifly as possible. Don't breathe heavily over them!
#### For lab on October 3
1. Check the data sheet for your primers. Add the indicated volume of 1X [TE buffer](https://en.wikipedia.org/wiki/TE_buffer#Recipe) to create a primer stock solution.
2. Vortex the solution for 10 secends.
3. Incubate the solution on the heat block at 55˚C for 5 min.
4. Vortex the solution for 10 secends.
5. Make a working primer solution by diluting the stock 1:10. Combine 95 μl of nuclease-free water with 5 μl of primer stock in a 0.5-ml tube. Vortex the solution. Label the tube.
#### PCR
6. Combine the following reagents in a 200-μl PCR tube:
| | |
| ------:|:------------------ |
| 6.0 μl | [JumpStart Taq Mix](https://www.sigmaaldrich.com/catalog/product/sigma/d9307) |
| 0.5 μl | gene-specifc primer |
| 0.5 μl | universal-Cas9sg-R primer |
| 5.5 μl | water (nuclease-free) |
| 12.5 μl | total |
7. Mix the reaction by pippetting up and down. (Set a pipetter to 3 μl, then gently pipette the solution up and down a few times. Don't intentionally remove any volume.)
8. Close the tube and label it with your group's initials.
9. Run a thermocycler program with the following conditions. (This should take about 45 minutes.)
| | | |
| ---- |:------ |:------------ |
| 98˚C | 10 s | \| |
| 60˚C | 30 s | \| 35 cycles |
| 72˚C | 15 s | \| |
| 72˚C | 10 min | |
| 12˚C | hold | |
#### RNA synthesis
Next we will use a modification of the MEGAscript T7 Transcription Kit (Life Technologies [item AM1334](https://www.thermofisher.com/order/catalog/product/AM1334)). The nucleotide triphosphates (NTPs) arrive in individual tubes. These should be combined into a new tube with equal volumes (and concentrations) of each.
10. Set up the T7 transcription reaction as follows in a 200-μl tube:
| | |
| ----:|:------------------- |
| 7 μl | nuclease-free water |
| 8 μl | NTPs |
| 2 μl | reaction buffer (10X) |
| 1 μl | template PCR product |
| 2 μl | T7 RNA polymerase enzyme mix |
| 20 μl | total |
11. Mix the reaction by gently pippetting up and down.
12. Incubate the reaction at 37˚C for 1-4 hours, using the incubation mode on the thermocycler. (A range of 1-16 hours seems to work. The optimum appears to be 4 hours.)
13. When the incubation is done, without cancelling the run, open the machine and remove the reaction tubes.
14. Add 1 μl of [TURBO DNase](https://www.thermofisher.com/order/catalog/product/AM2238), mix well, and return the tubes to incubation at 37˚C for 15 min.
15. Cancel the incubation and run the program "Anneal dsRNA", which denatures the RNA and inactivates the DNase by heating it, then cools slowly to allow the complementary RNA to adopt its proper secondary structure.
| | |
| ----:|:------------------- |
| 95˚C | 3 min |
| -0.1˚C/s ramp | takes about 9 min |
| 45˚C | 1 min |
| 12˚C | hold |
16. Add 79 μl of nuclease-free water.
17. Transfer the solution to a nuclease-free 0.5 ml tube.
#### RNA purification
18. Add 50 μl of 7.5M ammonium acetate and 300 μl 100% ethanol. Both these reagents should be stored at -20˚C until use.
19. Vortex the sample.
20. Precipitate the RNA at -20˚C for at least 20 min (or up to 2 days).
21. Centrifuge for 20 min at 12,000 rpm at 4˚C. Discard the supernatant.
22. Wash the pellet with 0.5 ml cold 70% ethanol.
23. Centrifuge for 5 min at 10,000 rpm at 4˚C. Discard the supernatant.
24. Dry the pellet in a vacuum centrifuge ("speed-vac") on the medium heat setting. Be sure to leave the caps open to allow evaporation. This should take about 10-15 minutes. Stop before the pellet becomes an opaque white color.
25. Resuspend the RNA pellet in 30 μl (suggested) of nuclease-free low Tris-EDTA Buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0)
26. Use the NanoDrop spec to measure the concentration and purity of the RNA. Record this information in your notebook. Concentrations should be at least 1 μg/μl, but may vary depending on the length of time allowed for RNA synthesis. Pure RNA should have an A~260~/A~280~ ratio greater than 2.0.
27. To confirm the quality of the RNA, run out 1 μl on a 2% agarose gel. It's convenient to run the RNA beside the template PCR product. (Be aware that single-strand RNA will not run to the same distance as double-strand DNA of the same length.)
28. Based on the measured concentration of RNA, dilute the solution to 500 ng/μl in nuclease-free low Tris-EDTA Buffer. (Use the old dilution equation, *c~1~v~1~ = c~2~v~2~*. Start with an initial volume, *v~1~*, that estimates the total volume of sgRNA solution available.)
29. Aliquot 5 μl into 0.5-ml tubes
30. Store at -80°C
#### RNA yields
Below are the RNA yeilds, purity (A~260~/A~280~ over 2.0 are pure RNA) from October 3, along with volumes for the preparation of a 500 ng/μl RNA solution.
| target | RNA (ng/μl) | A~260~/A~280~ | RNA (μl) | water (μl) | total (μl) |
|:-------- | -----:| -----:| ---:| ---:| ---:|
| EGFR-364 | 548.5 | 1.96 | 25 | 2.43 | 27.43 |
| EGFR-1041 | 2043.0 | 2.02 | 25 | 77.15 | 102.15 |
| Hpo-6 | 3032.5 | 2.05 | 25 | 126.63 | 151.63 |
| Hpo-7 | 2644.8 | 2.10 | 25 | 107.24 | 132.24 |
| optix | 1738.5 | 2.09 | 25 | 61.93 | 86.93 |
| spineless | 3649.0 | 1.90 | 35 | 220.43 | 255.43 |
| Wnt-354 | 761.3 | 1.71 | 25 | 13.07 | 38.07 |
| Wnt-712 | 654.7 | 1.67 | 25 | 7.74 | 32.74 |
Solutions of 500 ng/μl sgRNA were aliquoted into 5 μl and stored at -80˚C.
### Preparation of Cas9
The instructor will prepare this stock of Cas9 prior to lab.
#### As described by Martin et al.
1. Resuspend 65 μg NLS-Cas9 ([QB3 Berkeley](http://qb3.berkeley.edu/macrolab/cas9-nls-purified-protein/)) in 45 μl RNAse-free water.
2. Add 10 μl 0.5% [phenol red](https://en.wikipedia.org/wiki/Phenol_red) solution
3. Aliquot 5 μl into 0.5-ml tubes
4. Store at -80°C
#### Proedure used on 9 October
NLS-Cas9 ([QB3 Berkeley] arrived in 10 μl aliquots of a solution at 6.4 μg/μl. I diluted this to approximate the concentration specified above.
- 34.3 μl of nuclease-free water was added to one Cas9 aliquot.
- 9.8 μl of 0.5% phenol red was added.
- After mixing, 5 μl aliquots were placed in 0.5 ml tubes and stored at -80˚C.
### Prepare the sgRNA/Cas9 solution
1. Thaw 5 μl of Cas9 stock and 5 μl of sgRNA on ice
2. Combine and add 10 μl RNAse-free water
3. Mix gently by pipetting
4. Incubate at room temperature for about 10 min, then keep on ice until use
Final concentrations should be about 250 ng/μl Cas9, 125 ng/μl sgRNA, and 0.02% phenol red.
### Collecting eggs for injection
Two days after eclosure, adult butterflies should start mating and should produce fertile eggs for the next 10 days.
- About 4 hours before the time you plan to inject, place a few leaves of [hollyhock](https://en.wikipedia.org/wiki/Alcea) or [mallow](https://en.wikipedia.org/wiki/Malvaceae), or a stem of sunflower, in water, and position in the cage next to an incandescent light bulb to stimulate activity around the plant with light and heat.
- Females will drum on the plant before laying eggs. Note that time to brakcet the age of the eggs that are laid.
- Remove the eggs from the leaves to a small container. Do this with gloved hands or a soft clean paint brush.
- Add a flat strip of 3M/Scotch Brand double-sided tape to the inside of a small plastic cup lid. Decrease the stickiness by pressing the tape with a gloved finger. Dab it with a KimWipe to cover it with fibers. This will allow the hatchlings to walk away from the tape without getting stuck.
- Drop about 30 eggs directly on the tape. Reposition the eggs if needed using a soft paintbrush.
- Under a microscope, reposition eggs with the micropyle up using a brush or unpulled glass capillary. Press gently on top of the eggs to adhere them to the tape.
- Move to an injection station.
### *Vanessa* egg microinjection
- Take an injection needle (a pulled capillary tube made of borosilicate glass).
- Mount the needle on the micro-injector.
- Use the micromanipulator to position the needle in the center of the microscope view field.
- Break the tip off gently using clean forceps. Do so at the point where the pulled glass bends naturally.
- Remove the needle and back-load it with 1 μL of injection mix. Then return it to the micro-injector.
- Press the foot pedal to release a small amount of fluid.
- Move the lid with one hand so the needle is at a 45° vertical angle, slightly above the egg top. Focus the microscope so the egg top is sharp.
- Start with a pressure of 20 psi and keep the injection diameter minimal. Change the needle or reduce pressure down to 10 psi at anytime during the injection if expelled droplets become more than twice the size of the micropyle.
- Holding the egg-covered lid firmly with one hand, use the coarse knob to puncture the upper side of the egg. Do not go very deep beyond the chorion. Release a minimal volume of sgRNA/Cas9 solution. Backflow of yolk will occasionally occur, but is often expelled by repeated bursts.
- While still holding the egg dish stedily, retract the needle and inject outside of the egg to expel any yolk. If nothing comes out, the needle will usually unclog after puncturing the next egg. Try to achieve consistency and decent speed.
- Upon piercing the egg, only add enough solution so that when you pull the need out you can clearly see the red solution inside the egg.
- Once all the eggs have been injected, count them, record information on the lid, such as the name of the person making the injections, the date, the age range of the embryos, and the target sgRNA.
- Snap the lid to a matching cup
- Place the cups in a closed plastic conatiner with a wet paper towel to avoid desiccation during the next 24 hrs. Include a control batch of uninjected eggs in a similarly prepared dish.
- Open the tupperware after 24 hrs to prevent condensation.
### Care of experimental animals
- Embryos will develop at a temperature-dependent rate, hatching after about 3 days at 28°C to about 4 days at 22°C. In the morning of the hatching day, press small crumbs of fresh artifical larval diet onto the side and lid of the cup. Hatchlings should be found feeding on the diet within 24 hrs.
- Record the number of hatchlings. Typically 30-40% of injected eggs survive for WntA sgRNA/Cas9 injections made by an experimented operator. Low rates of survival may indicate lethal phenotypes, in which case you can inject older eggs, or with a more dilute sgRNA/Cas9 solution in order to generate mosaic embryos with fewer editted cells.
- Leave the larvae at room temperature (22-25°C). Check for excess moisture daily. Temperatures of 24-25°C (75°F) provide an optimal trade-off between developmental time and survival, with a developmental time of 30-36 days. Pinholes in the lid may be necessary to let excess moisture out of the cup.
- Examine the larvae every two days.
- Add small amounts of fresh diet as needed.
- Remove individuals to seperate cups once they reach the third instar.
- Dispose of dead larvae as biohazardous waste.
- Hang pupae in a dedicated cage. Spray them with water every 1-2 days. Record the number of pupae. Note any abnormalities.
- After emergence, allow 1 day for wings to fully to dry, then freeze all G~0~ adults at -20°C for >24hrs.
- Pinning is not necessary for this class and phenotypic analysis can be done on wings that have been detached from the insect body and stored in glassine envelopes.
## Results - so far
#### 15 October
After injections on Thursday, embryos were left to develop at room temperature (roughly 22˚C) over the weekend. On Monday, nothing had hatched. Tuesday (October 15) several hatchlings had emerged. These treatments were moved to the incubator at 25˚C and given about 0.5 ml of artificial larval diet.
Importantly, it looks like many of the eggs, even in control treatments, did not begin development at all. (They still had the uniform light green color of freshly laid eggs.) These may have been unfertilized eggs. The overall average embryo survival for the control treatments was about 48%.
Most of the CRISPR treatments have at least a few hatchlings. These may be viable editted animals, however it's possible these are "escapers," individuals who were not actually injected.
| lab group | sgRNA | eggs collected | embryos developed | hatchlings | embryo survival |
|:------:|:----------:|:----:|:----:|:----:|:----:|
| AR, MK | uninjected | 15 | 4 | 4 | 26.7% |
| MR, CR | uninjected | 13 | 11 | 8 | 61.5% - 1 dead by 10/22 |
| JW, BS | uninjected | 5 | 3 | 0 | 0 |
| JW, BS | uninjected | 4 | 4 | 3 | 75.0% |
| JW,BS | EGFR-364 | 15 | 0 | 0 | 0 |
| JW,BS | EGFR-1041 | 5 | 3 | 0 | 0 |
| Dr.A. | EGFR-1041 | 59 | 4 | 1 | 1.7% |
| Dr.A. | EGFR-1041 | 29 | 5 | 2 | 6.9% |
| MR, CR | Hpo-6 | 20 | 1 | 1 | 5% |
| TM, YS | optix-? | 8 | 1 | 1 | 12.5% -- died by 10/22 |
| AR, MK | ss-? | 9 | 0 | 0 | 0 |
| EL, BM | Wnt-354 | 7 | 2? | 1 | 14.3% |
| EL, BM | Wnt-712 | 7 | 2? | 1 | 14.3% -- died by 10/22 |
## Materials
### Animals
- *Vanessa cardui* caterpillars (nature-gifts.com, [Caterpillar Refill Kit](https://www.nature-gifts.com/shop/grow-butterflies/caterpillar-refill-kit/))
### Larval food
This recipe is basd on instructions from Frontier Agricultural Sciences and Martin et al. (in press). Item #F9698B from Frontier Agricultural includes a dry mix (including soy flour, wheat germ, brewer's yeast, sugars, salts and perservatives), KOH solution, and dry agar. (Enough to make 10L.) The instructions below are for preparation of 1L. Ingredients can be mixed using a handheld kitchen mixer.
- In a 2-L beaker, mix 19 g dry agar with 710 ml water
- On a hot plate with a large stirring bar, bring the solution to a boil for 1 min
- Remove the mixture from the heat
- Add 246.1 g of dry diet and mix with hand-held mixer
- Add 6.84 ml of KOH solution (provided with the Frontier Agricultural diet) and mix for 10 s
- Add 300 ul of 25% (v/v) glacial acetic acid and mix for 10 s
- Optionally, add up to 30 g of fresh leaves from mallow, hollyhocks or sunflower
- Blend for 20 s or until thoroughly mixed
- Add 4 ml formaldehyde (37%)
- Add 8 ml canola oil
- Blend for 30 s or until thoroughly mixed
- Aliquot into 1.25 oz Plastic Souffle Cups (Frontier Agriculture Sciences, item 9091)
- Store in a sealed container at 4˚C for up to 2 weeks
### Housing and equipment for raising caterpillars
- an incubator
- 1.25 oz Plastic Souffle Cups (Frontier Agriculture Sciences, [item 9091](https://insectrearing.com/product/1-25-oz-plastic-cup/))
- tray for cups (Frontier Agriculture Sciences, [item 9040](https://insectrearing.com/product/cup-tray-30-wells/))
Pack larval food into the bottom of the cups. Each caterpillar needs 8-10 ml of food to complete development. Cover the cup with a small square of paper towel or KimWipe and snap on the lid. Keep them at 23-25°C with 40-60% relative humidity and a 12/12 light/dark cycle. Cover them from direct light to minimize condensation.
### Housing and equipment for keeping adult butterflies
- collapsible mesh enclosure (Carolina Biological Supplies, [item #674291](https://www.carolina.com/butterfly-habitats/carolina-butterfly-sanctuary/674291.pr))
- fresh leaves of mallow, hollyhock, or sunflower (available from a florist) in a cup or beaker of water
- an incandescent light bulb to heat plants to encourage oviposition
- a 1:1 solution of Gatorade and spring water
- a feeding station can be purchased with butterfly kits (e.g. from nature-gifts.com) or constructed by cutting a hole in a small plastic container and creating a wick out of a cotton ball
## References
- **Barrangou R.** (2015). The roles of CRISPR-Cas systems in adaptive immunity and beyond. *Curr. Opin. Immunol*. **32**, 36-41. [Link](https://www.ncbi.nlm.nih.gov/pubmed/25574773).
- **Beldade P, Brakefield PM**. (2002). The genetics and evo-devo of butterfly wing patterns. *Nature Reviews Genetics*. **3**(6), 442-52. [Link](https://www.ncbi.nlm.nih.gov/pubmed/12042771)
- **Connahs, H., Tlili, S., van Creij, J., Loo, T. Y., Banerjee, T. D., Saunders, T. E., Monteiro, A.** (2019). Activation of butterfly eyespots by *Distal-less* is consistent with a reaction-diffusion process. *Development*. **146**, dev169367. [Link](https://dev.biologists.org/content/146/9/dev169367)
- **Martin A, Wolcott NS, O'Connell LA.** (in press). Bringing immersive science to laboratory courses using CRISPR gene knockouts in butterflies and frogs. *J. Evol. Biol.*
- **Mazo-Vargas, A., Concha, C., Livraghi, L., Massardo, D., Wallbank, R. W., Zhang, L., Papador, J. D., Martinez-Najera, D., Jiggins, C. D., Kronforst, M. R.** (2017). Macroevolutionary shifts of WntA function potentiate butterfly wing-pattern diversity. *Proc. Natl. Acad. Sci.* **114** (40), 10701-10706. [Link](https://www.pnas.org/content/114/40/10701)
- **Zhang, L. & Reed, R. D.** (2016). Genome editing in butterflies reveals that *spalt* promotes and *Distal-less* represses eyespot colour patterns. *Nature Communications*. **7**, 11769. [Link](https://www.nature.com/articles/ncomms11769)
- **Zhang, L. & Reed, R. D.** (2017). A Practical Guide to CRISPR/Cas9 Genome Editing In Lepidoptera. In *Diversity and Evolution of Butterfly Wing Patterns: An Integrative Approach* (eds. Sekimura, T. & Nijhout, H. F.), pp.155–172. Singapore: Springer Singapore. [Link](https://www.researchgate.net/publication/319369908_A_Practical_Guide_to_CRISPRCas9_Genome_Editing_in_Lepidoptera/fulltext/59a74da3a6fdcc61fcfbd675/A-Practical-Guide-to-CRISPR-Cas9-Genome-Editing-in-Lepidoptera.pdf)
- **Zhang, L., Mazo-Vargas, A. and Reed, R. D.** (2017a). Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. *Proc. Natl. Acad. Sci.* **114**, 10707–10712. [Link](https://www.pnas.org/content/114/40/10707)
- **Zhang, L., Martin, A., Perry, M. W., van der Burg, K. R. L., Matsuoka, Y., Monteiro, A., Reed, R. D.** (2017b). Genetic basis of melanin pigmentation in butterfly wings. *Genetics*. **205**(4), 1537-1550. [Link](https://www.genetics.org/content/205/4/1537)
- **Zuo Z, Liu J.** (2016). Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. *Scientific Reports*. **6**, 37584. [Link](https://www.nature.com/articles/srep37584)