# OTU 與 ASV 的差別
###### tags: `bioinformatics`
閱讀 metabarcoding(高通量分子條碼)的研究時,因為會定序出多量短片段的序列需要分析,常會看到 OTU 或者 ASV 這類名詞。
OTU 是操作分類單元(operational taxonomic units),而 ASV 是擴增子序列變異(amplicon sequence variant)。OTU 的概念很早以前就出現[^2],而 ASV 的概念一直到 2013 年才被提出[^1];雖然中間或後續也有許多名詞如 ZOTU(zero-radius OTU)、ESV (Exact Sequence Variant)、sOTU (sub-OTU)、ISU (individual sequence variant),不過概念均與 ASV 相當類似,因此下文就主要比較 OTU 與 ASV 的差別。
### 兩者的不同
OTU 與 ASV 的差別在演算的方式,前者用的是聚類(clustering),後者用的是序列校正或者去噪(denoising)。
OTU 的產生,主要是據序列的相似性,以一定的相似度把序列歸集到一起。一般用的相似度是 97% 或 99%,產生出來的 OTU 就叫 OTU97 或 OTU99。
以 NGS 產出的資料來說,這個方法的缺點主要是敏感度不足,一個 OTU 中可能包含有不同的實際物種;再者,如果序列因為定序過程產生錯誤,會造成不可信賴的聚類結果。目前比較多人用以產出 OTU 的工具,是 [USEARCH](https://drive5.com/usearch/) 中的 UPARSE 演算法。USEARCH 本身為付費軟體,不過有開放 32 位元版免費使用。

從特定區段序列擴增到 OTU 產生的示意圖,取自[維基百科](https://en.wikipedia.org/wiki/Amplicon_sequence_variant)。
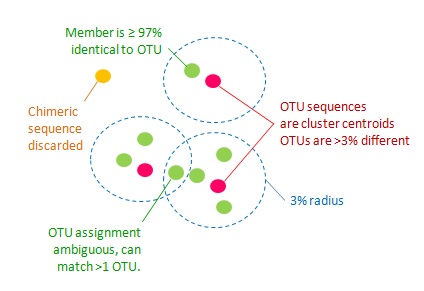
取自 [UPARSE-OTU algorithm](https://drive5.com/usearch/manual/uparseotu_algo.html) 的解說網頁
ASV 的取得,主要是針對定序時的錯誤來做修正。以目前最主流的 DADA2 演算法來說,就是根據 Illumina 定序的錯誤模型,藉由演算法與非監督是學習方法 selfConsist 來去除因為定序所造成的序列錯誤,而產出 ASV。因此相較於 OTU,ASV 的區分精細度就比較高。

從特定區段序列擴增到 ASV 產生的示意圖,取自[維基百科](https://en.wikipedia.org/wiki/Amplicon_sequence_variant)。注意與 OTU 的不同。
### 不同的 ASV 演算法之爭
2013 年提出的 UPARSE 演算法[^5]已經大大提高了 OTU 的精確程度,之後 2016 年 DADA2 發表[^3],文中說這套工具的演算法比 UPARSE 還精確,就引燃了戰火。UPARSE 的提出者 Robert C. Edgar 馬上在三個月後發表了一套去噪工具 UNOISE2,摘要只有三行,就為了宣稱 UNOISE2 產生 ASV 的效率與效果比 DADA2 還好[^4]
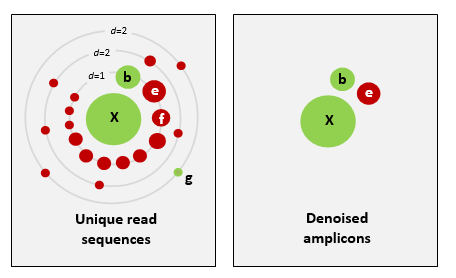
UNOISE2 的演算法[原理簡介](http://www.drive5.com/usearch/manual/unoise_algo.html)。
之後,Edgar 又出了 UNOISE3,包含於他的 USEARCH 軟體之內,又將 UNOISE3 產出的結果稱為 ZOTU ~~徒增名詞造成大家溝通的困擾~~。USEARCH 的特點就是以 C 語言寫成,處理大量數據的效率比 R 好(DADA2 一開始是以 R 套件包的形式發布),不過因為未開源,64 位元版本需要購買,目前還是滿多人使用 DADA2 系列演算法的。
### 其他演算法
除了前面提到的 USEARCH、UNOISE 系列演算法與 DADA2 之外,還有其他用來從定序資料中提取相異序列的方法,如 2017 年的 Deblur[^6] 與 2020 年發表的 AmpliCI[^7]。另外還有一些從頭聚類(*de novo* clustering)的方法,就比較少人使用了。本篇主要介紹 metabarcoding 最主流的 OTU 與 ASV 以及相關變體的區別;了解區別之後,就可以開始選擇合用的演算法工具進行分析了。
<span style="font-size:30px">🐕🦺</span>2022.12.17
[^1]: Eren, A. M., Maignien, L., Sul, W. J., Murphy, L. G., Grim, S. L., Morrison, H. G., & Sogin, M. L. (2013). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. *Methods in ecology and evolution*, *4*(12), 1111-1119.
[^2]: Sokal & Sneath: *Principles of Numerical Taxonomy*, San Francisco: W.H. Freeman, 1963
[^3]: Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., & Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. *Nature methods*, *13*(7), 581-583.
[^4]: Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. *BioRxiv*, 081257.
[^5]: Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. *Nature methods*, *10*(10), 996-998.
[^6]: Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Zech Xu, Z., ... & Knight, R. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. *MSystems*, *2*(2), e00191-16.
[^7]: Peng, X., & Dorman, K. S. (2020). AmpliCI: a high-resolution model-based approach for denoising Illumina amplicon data. *Bioinformatics*, *36*(21), 5151-5158.